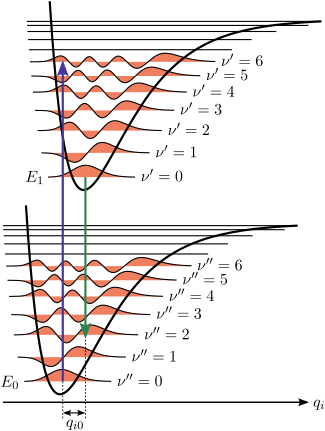
 圖1.
弗蘭克-康登原理能量圖,因?電子躍遷相對於核運動是瞬間的,在核座標下,振動能級趨向最小變化,這個例子指出位能井中,v = 0 和 v = 2之間最有可能的躍遷。
圖1.
弗蘭克-康登原理能量圖,因?電子躍遷相對於核運動是瞬間的,在核座標下,振動能級趨向最小變化,這個例子指出位能井中,v = 0 和 v = 2之間最有可能的躍遷。
弗?克-康登原理
(
英語:
Franck?Condon principle
)是
光??
的重要原理,用于解??子-振???的强度。?子-振???指分子吸收或?射光子后,?子能?和振?能?同??生?化的?程。弗?克-康登原理指出,在
分子?子??
?程中,???振?能?(分??于不同的?子能?)的
波函?
有效重?程度最大?,???振?能?之?的???生的?率最大。此原理可以被
量子力?
所解?。
 圖2.
此?在圖一能量圖中,吸收光譜與螢光光譜的圖示。因?基態與激發態的位能井形狀相同,所以造成光譜的對稱性。細線的部分通常僅在稀釋的氣體光譜中被觀察到。比較暗的曲線代表相同躍遷在液體?氣體中的非均勻致寬。在吸收光譜及螢光光譜中,最低振動能級之間躍遷(0?0 躍遷)有相同的能量。
圖2.
此?在圖一能量圖中,吸收光譜與螢光光譜的圖示。因?基態與激發態的位能井形狀相同,所以造成光譜的對稱性。細線的部分通常僅在稀釋的氣體光譜中被觀察到。比較暗的曲線代表相同躍遷在液體?氣體中的非均勻致寬。在吸收光譜及螢光光譜中,最低振動能級之間躍遷(0?0 躍遷)有相同的能量。
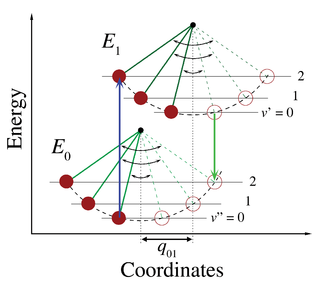 圖3.
弗蘭克-康登原理的半經典單擺類比。因?動量與核座標在兩個代表能級上相符,因此振動能級躍遷會發生在轉折點。本圖例中,0?2電子-振動躍遷較受??。
圖3.
弗蘭克-康登原理的半經典單擺類比。因?動量與核座標在兩個代表能級上相符,因此振動能級躍遷會發生在轉折點。本圖例中,0?2電子-振動躍遷較受??。
?述
[
??
]
弗蘭克 - 康登原則是成熟的半經典解釋,由
詹姆斯·弗?克
提出
[1]
。因?電子躍遷與核運動的時間尺度相比基本上是瞬時的,因此分子如果在電子躍遷期間移動到新的振動能級,這個新振動能級的核位置和動量須立?與原電子態的分子振動能級兼容。在簡諧振?器的半經典圖像中,必要條件發生在動量?零的轉折點處。
經典上,弗蘭克 - 康登原理是個近似:電子躍遷最可能發生在分子實體及其環境中原子核位置不變的情況下。由此?生的態被稱?弗蘭克 - 康登態,且垂直躍遷參與其中。該原理的量子力學公式?「振動躍遷的?度,與躍遷中涉及的兩種態的振動波函數的重疊積分平方成正比」
-? IUPAC化學術語彙編,第2版(1997年)
振動能級和振動波函數是
量子諧振子
的函數,或是更複雜的近似分子勢能,如
莫?斯?
。圖1?具有莫爾斯勢能函數的分子,基態及激發態的振動躍遷,說明了弗蘭克-康登原理。在低溫近似中,分子從一開始的電子基態及振動能級
v
= 0,吸收必要能量的光子後,躍遷?電子激發態。新態的電子排布可能導致構成分子的原子核的平衡位置發生位移。在圖1中,基態和第一激發態之間的核坐標位移記作
q
01
。若考慮最簡單的雙原子分子情形,則核坐標軸指的是核間分離。因?躍遷過程中假定核坐標不變,所以振動躍遷用垂直箭頭表示。分子最終到達任何特定振動能級的?率,將與原始態和最終態振動波函數(垂直)重疊的平方成正比(參見下面的量子力學公式部分)。在電子激發態下,分子迅速弛豫到最低電子激發態的最低振動能級(
?莎??
),?從那里發射光子而衰減到電子基態。弗蘭克-康登原理同樣適用於吸收和熒光。
弗蘭克-康登原理在吸收和熒光方面的適用性以及?莎規則,導致了如圖2所示的近似鏡像對稱性。由於沒有個別躍遷的不均勻加寬,分子在冷而稀疏的氣體中的振動結構最?明顯。圖2中所示的振動躍遷是窄的等間距洛倫?線。振動能級之間的等間距僅適用於簡諧振子的?物線勢。在如圖1所示,更?實的勢中,能量間距隨振動能量的增加而減小。從最低振動態到最低振動態的電子躍遷通常被稱?0-0(zero - zero)躍遷,?且在吸收和熒光中具有相同的能量。
發展
[
??
]
在《法拉第學會學報》1926年發表的一篇報告中,詹姆斯·弗蘭克對光子引發的化學反應機制進行了?究。假設的機制是一個光子激發一個分子,接著在短暫的激發時間內與?一個分子發生?撞。現在問題是「一個分子是否可能在不發生?撞的情況下,通過光子的吸收,一個步驟就分解成光?物。」?了使分子分解,?必須從光子中獲得超過解離能的振動能,?打破化學鍵的能量。然而,正如當時所知,分子只吸收與允許量子躍遷相對應的能量,在勢?的解離能級之上沒有振動能級。若吸收高能光子,則會向更高的電子態躍遷而不是解離。?了?究分子被激發到高電子能級時能獲得多少振動能,以及這種振動能量是否足以立?將分子分解。他?了三個圖表,表示最低電子態和最高電子態之間結合能的可能變化。
圖I中顯示了從正常態n到激發態a和a'躍遷過程中,束縛大幅地減弱。這裡有D' > D'和D' > D" ,同時,原子核的平衡位置隨著激發移動到更大的r?。如果我們從n曲線的平衡位置(最小勢能)
垂直
向上躍遷到圖I中的曲線,粒子將具有大於D'的勢能,?將飛離而去。在這種情況下,我們在光激發時的振?能量有?大的變化。
??詹姆斯·弗蘭克,1926年
詹姆斯·弗蘭克認識到,振動能級的變化可能是電子能級激發的瞬時性質以及核交互作用勢的新平衡位置的結果。愛德華·康登在1926年發表的一篇名?《能帶系統?度分佈理論》的物理評論文章中
[2]
,將這一觀點擴展到了光反應之外。在這裡,他以與現代形式非常相似的方式闡述了半經典的表述。關於這個新原理,弗蘭克和康登的第一次聯合引用出現在1926年同一期的《物理評論》(Physical Review)一篇關於一?化?能帶結構的文章中。由雷蒙德·伯奇(Raymond Birge)所發表。
 圖4.
愛德華·康登關於現在的弗蘭克-康登原則的第一?出版物[康登1926]。康登選擇疊加勢曲線來說明?算振動躍遷的方法。
圖4.
愛德華·康登關於現在的弗蘭克-康登原則的第一?出版物[康登1926]。康登選擇疊加勢曲線來說明?算振動躍遷的方法。
量子力學公式
[
??
]
考慮一個
?偶?矩
,從基態電子能級 (ε) 的初始振動能級 (υ)「
 」(見
狄拉克符?
),躍遷到激發態 (ε′) 的振動能級 (υ′)「
」(見
狄拉克符?
),躍遷到激發態 (ε′) 的振動能級 (υ′)「
 」,分子偶極算符
μ
由電子的電荷 (?
e
)及位置 (
r
i
) 和核的電荷 (+
Z
j
e
) 和位置 (
R
j
) 決定。
」,分子偶極算符
μ
由電子的電荷 (?
e
)及位置 (
r
i
) 和核的電荷 (+
Z
j
e
) 和位置 (
R
j
) 決定。

兩種態之間躍遷的?率幅
P
由下式給出

 及
及
 分別是初始態及最終態的總體波函數。總體波函數是個體振動波函數(取決於原子核的空間坐標)、電子空間波函數、電子自旋波函數的乘積:
分別是初始態及最終態的總體波函數。總體波函數是個體振動波函數(取決於原子核的空間坐標)、電子空間波函數、電子自旋波函數的乘積:

電子及振動波函數的分離利用了
?恩-?本海默近似
,這也是弗蘭克-康登原理的基本假設。 將這些方程組合在一起,可以根據單獨電子空間,自旋及振動的貢獻來表達?率幅:



原本的積分中與自旋無關的部分,近似?兩個積分的乘積:

如果電子空間坐標上的積分
 不依賴於核坐標,則這個分解才會正確。 然而在?恩?奧本海默近似中
不依賴於核坐標,則這個分解才會正確。 然而在?恩?奧本海默近似中
 及
及
 與核坐標相依, 以致於這個積分,也就是所謂的躍遷偶極表面 (
transition dipole surface
) 是一個核坐標函數。 這個相依性通常因??平滑而被忽略(也就是通常假設
躍遷偶極表面
與核坐標無關,稱?康登近似。)
與核坐標相依, 以致於這個積分,也就是所謂的躍遷偶極表面 (
transition dipole surface
) 是一個核坐標函數。 這個相依性通常因??平滑而被忽略(也就是通常假設
躍遷偶極表面
與核坐標無關,稱?康登近似。)
因?不同態的電子波函數是正交的,所以加號後的第一個積分等於零。 剩下的部分是三個積分的乘積。 第一個積分是振動重疊積分,也稱?
弗蘭克-康登因子
。而剩餘對?率幅有貢獻的兩個積分,則決定了電子的軌道選擇規則與自旋選擇規則。
弗蘭克-康登原理陳述了兩個不同電子態之間所允許的振動躍遷; 而其他量子力學選擇規則可以降低躍遷?率,或完全禁止躍遷。 上述推導中忽略了旋轉選擇規則。旋轉的貢獻可以在氣體光譜中被觀察到,但在液體及固體中,旋轉貢獻被?烈抑制。
應?楚了解的是,弗蘭克-康登原理的量子力學公式是一系列近似的結果,主要是電偶極躍遷假設及?恩?奧本海默近似。因?總體波函數不能完全有效分解?核波函數、電子空間波函數、電子自旋波函數,意味著較弱的磁偶極子及電四極電子躍遷不會嚴格地遵守選擇規則(包括弗蘭克-康登因子)。對於任意給定的躍遷,
P
的?由所有選擇規則來決定,但是最大的貢獻來自於自旋選擇規則,其次是電子選擇規則。 弗蘭克-康登因子僅微弱地調製躍遷?度,也就是,?對能帶?度的因子大約?1,能帶?度數量級取決於其他選擇規則。下表?可能組合的吸收係數範圍,包含允許及禁止、自旋及軌道的選擇規則。
電子躍遷?度
|
莫耳吸光度
(ε) ?範圍 (
mol
?1
cm
?1
)
|
| 自旋及軌道皆允許
|
10
3
到 10
5
|
| 自旋允許但軌道禁止
|
10
0
到 10
3
|
| 自旋禁止但軌道允許
|
10
?5
到 10
0
|
弗蘭克-康登原理在光譜學中的比?
[
??
]
 圖5
.沿著位形坐標
q
i
,與聲子?合的電子躍遷能量圖,這是晶格的
?正模
。 向上箭頭表示沒有聲子和吸收三個聲子的情形。向下箭頭則表示發射的對稱過程。
圖5
.沿著位形坐標
q
i
,與聲子?合的電子躍遷能量圖,這是晶格的
?正模
。 向上箭頭表示沒有聲子和吸收三個聲子的情形。向下箭頭則表示發射的對稱過程。
在弗蘭克-康登原理的規範型式裡,電子吸收或發射光子的過程中只採用分子振動能級的變化,在物理直覺上,在短?的電子躍遷過程中,構成分子的原子核座標來不及改變,而弗蘭克-康登原理也是基於這個想法。這種物理直覺可以常規地擴展到光吸收或發光分子(
發色團
)與其環境之間的交互作用。這是因?分子經常與周圍分子?烈交互作用(特別是在液體及固體中),而這些交互作用以非常類似於弗蘭克-康登原理所考慮的分子振動的方式,改變發色團的核坐標,因此弗蘭克-康登比?是合理的。
聲子的弗蘭克-康登原理
最接近的弗蘭克-康登類比:「發色團作?雜質嵌入晶格中,而
?子
(晶格振動的量子)會與發色團的電子躍遷?生交互作用。」在這種情況下,當光子的能量對應於純電子躍遷能量,或對應於純電子躍遷能量加上一個或多個晶格聲子的能量時,就會發生躍遷到更高的電子能級。在低溫近似中,發射躍遷是從激發態的零聲子能級到基態的零聲子能級或基態的高聲子能級。涉及聲子的躍遷?率是由聲子波函數在初始能級和最終能級上的重疊決定的,這與弗蘭克-康登原理的情形一致。對於應用在聲子躍遷的弗蘭克-康登原理,圖1中水平軸的標籤在圖5中用
?正模
的位形坐標取代。圖5中晶格模式
 的勢能表示?諧振子的勢能,而聲子能級之間的間距(
的勢能表示?諧振子的勢能,而聲子能級之間的間距(
 )由晶格參數確定。因?單個聲子的能量通常非常小,所以只能在低於約40 開爾文的溫度下觀察到零或幾個聲子躍遷。
)由晶格參數確定。因?單個聲子的能量通常非常小,所以只能在低於約40 開爾文的溫度下觀察到零或幾個聲子躍遷。
- 有關更多詳細信息和參考,請參見
零聲子線和聲子邊帶
。
溶劑化中的弗蘭克-康登原理
當發色團溶於液體,其電子躍遷也可以應用弗蘭克-康登原理。在這個弗蘭克-康登比?中,發色團振動能級還有發色團-液體聲子的交互作用,持續促成吸收和發射的光譜結構,但應將這些獨立效分開考慮。
當發色團被溶劑分子所包圍,尤其溶劑分子是極性的時候,周圍分子會與發色團發生交互作用。這種溶劑和溶質之間的關聯稱?
溶劑化
,而溶劑化是個穩定的交互作用,也就是溶劑分子會移動和旋轉,直到交互作用的能量達到最小化。交互作用會涉及到靜電和范德華力,也可以涉及?鍵。當交互作用在電子基態及激發態中不同時,就可以應用弗蘭克-康登原理。例如,交互作用的變化可能源於這兩種態的偶極矩不相同。如果發色團一開始在基態,?且與周圍的溶劑分子接近平衡,然後吸收一個光子後達到激發態,那??與溶劑的交互作用將遠離激發態的平衡。而在溶劑化情況下,溶劑分子會重新排列,這現象類似最初的弗蘭克-康登原理:「與核的運動相比,電子躍遷非常快 。」現在我們來說明垂直躍遷,但在這個例子裡,水平坐標是溶劑 - 溶質交互作用空間。該坐標軸抽像地表示了全部交互作用的溶劑分子運動的所有相關維度,通常標記?「溶劑化坐標」(Solvation Coordinate)。
最初的弗蘭克-康登原理中,電子躍遷之後,處於較高振動態的分子立?弛豫至最低振動態。而在溶劑化的情況下,溶劑分子將立?重新排列,使交互作用能最小化。此外溶劑弛豫的速率取決於
溶劑的粘度
。假設與電子激發態的壽命相比,溶劑弛豫時間較?短暫,那?發射躍遷將來自激發電子態的最低溶劑能態。對於環境溫度下的小分子溶劑如水或甲醇,溶劑弛豫時間約?幾十皮秒而發色團激發態的壽命範圍從幾皮秒到幾納秒。躍遷到基態電子態之後,溶劑分子必須立?重新排列,以適應發色團新的電子組態。圖6說明了應用於溶劑化的弗蘭克-康登原理。當溶液被與電子躍遷能相對應的光照射時,一些發色團會移動到激發態。如圖所示,在這組發色團中,溶劑-發色團交互作用能的統計分佈?高斯分佈函數。在兩種電子態中,溶劑 - 發色團交互作用被繪製??物線勢。由於電子躍遷在溶劑運動的時間尺度(垂直箭頭)上基本上是瞬時的,因此激發態發色團的聚集立?遠離平衡。接著如圖6中的曲線箭頭所示,溶劑分子會按照新的勢能曲線重新排列。注意,雖然電子躍遷是量子化的,但由於大量的電子躍遷,色譜 - 溶劑交互作用能仍被視?經典的連續譜。雖然發射發生在激發態色子-溶劑交互作用勢的最小?,但當溶劑粘度高或激發態壽命短時,仍可以在達到平衡之前發生顯著的發射。圖6所示的吸收光子和發射光子之間的能量差,是溶劑化對
斯托克斯位移
的貢獻。
 圖6.
能量圖說明了應用於發色團溶劑化的弗蘭克-康登原理。 ?物線電位曲線表示發色團和溶劑之間的交互作用能。 高斯曲線表示該交互作用能的分佈。
圖6.
能量圖說明了應用於發色團溶劑化的弗蘭克-康登原理。 ?物線電位曲線表示發色團和溶劑之間的交互作用能。 高斯曲線表示該交互作用能的分佈。
相??目
[
??
]
延伸閱讀
[
??
]
Franck, J. (1926). "Elementary processes of photochemical reactions". Transactions of the Faraday Society. 21: 536?542. doi:10.1039/tf9262100536. Link
Condon, E. (1926). "A theory of intensity distribution in band systems (Meeting abstract)". Physical Review. 27: 640. Bibcode:1926PhRv...27..637.. doi:10.1103/PhysRev.27.637.
Condon, E. (1926). "A theory of intensity distribution in band systems". Physical Review. 28: 1182?1201. Bibcode:1926PhRv...28.1182C. doi:10.1103/PhysRev.28.1182. Link
Condon, E. (1928). "Nuclear motions associated with electron transitions in diatomic molecules". Physical Review. 32: 858?872. Bibcode:1928PhRv...32..858C. doi:10.1103/PhysRev.32.858. Link
Birge, R. T. (1926). "The band spectra of carbon monoxide". Physical Review. 28: 1157?1181. Bibcode:1926PhRv...28.1157B. doi:10.1103/PhysRev.28.1157. Link
Noyes, W. A. (1933). "The correlation of spectroscopy and photochemistry". Reviews of Modern Physics. 5: 280?287. Bibcode:1933RvMP....5..280N. doi:10.1103/RevModPhys.5.280. Link
Coolidge, A. S, James, H. M. and Present, R. D. (1936). "A study of the Franck?Condon Principle". Journal of Chemical Physics. 4: 193?211. Bibcode:1936JChPh...4..193C. doi:10.1063/1.1749818. Link
Herzberg, Gerhard (1971). The spectra and structures of simple free radicals. New York: Dover. ISBN 0-486-65821-X.
Harris, Daniel C.; Michael D. Bertolucci (1978). Symmetry and spectroscopy. New York: Dover. ISBN 0-486-66144-X.
Bernath, Peter F. (1995). Spectra of Atoms and Molecules (Topics in Physical Chemistry). Oxford: Oxford University Press. ISBN 0-19-507598-6.
Atkins, P. W.; R. S. Friedman (1999). Molecular Quantum Mechanics. Oxford: Oxford University Press. ISBN 0-19-855947-X.
參考文獻
[
??
]
- ^
Franck, J(1926).
"
Elementary processes of photochemical reactions
"
. Transactions of the Faraday Society: 536-542.
doi:10.1039/tf9262100536
.
- ^
Condon, Edward.
A Theory of Intensity Distribution in Band Systems
. Physical Review. 1926-12-01,
28
(6): 1182?1201.
Bibcode:1926PhRv...28.1182C
.
doi:10.1103/PhysRev.28.1182
.