Синдром Марфана
?
генетичне
порушення розвитку
сполучно? тканини
.
[5]
Ступ?нь порушення вар?ю?. Хвор? на синдром Марфана зазвичай ? високими, худими, з довгими руками, ногами, пальцями ? ступнями, також для них характерн? надзвичайно гнучк? суглоби ?
скол?оз
.
[5]
Найсерйозн?ш? ускладнення зустр?чаються з боку
аорти
?
м?трального клапана
? у хворих часто д?агностують
аневризму аорти
та
недостатн?сть м?трального клапана
через пролапс його задньо? стулки.
[5]
[6]
?нш? органи, в яких можуть виникати зм?ни, включають леген?, оч?, к?стки та хребет.
[5]
Синдром Марфана ? аутосомно-дом?нантне захворювання. Приблизно у 75 % випадк?в воно ? спадковим, а решта 25 % спричинен? випадковими
мутац?ями
.
[5]
Мутац?я зазвичай виника? у ген?, який м?стить ?нформац?ю про синтез ф?брил?ну-1, що спричиню? порушення структури сполучно? тканини.
[5]
Д?агноз про наявн?сть синдрому Марфана у хворого часто базу?ться на використанн? критер??в Гента.
Нема?
ет?олог?чного
л?кування синдрому Марфана. Б?льш?сть хворих живуть звичайно, використовуючи ситуативне л?кування.
[5]
Воно часто включа? використання
бета-блокатор?в
, таких як
пропранолол
або, у раз? наявно? сенсиб?л?зац?? до нього,
блокатор?в кальц??вих канал?в
та ACE ?нг?б?тор?в.
[7]
[8]
Х?рург?чне втручання може бути необх?дне для корекц?? аневризми аорти та м?трального клапана.
[8]
До загальних рекомендац?й для хворих на синдром Марфана в?дносять уникнення виконання важких ф?зичних вправ.
[7]
Зустр?ча?ться р?дко ? 1:3.000-10.000 в?д загально? к?лькост? новонароджених мають синдром Марфана.
[7]
[9]
Зустр?ча?ться однаково часто як серед чолов?к?в, так ? серед ж?нок, як ? вс? автосомно-дом?нантн? захворювання.
[7]
Частота виникнення не пов'язана з расою ? з рег?оном.
[9]
Синдром Марфана був названий на честь
Антуана Марфана
,
[10]
французького пед?атра, який уперше описав цей стан у 1896 роц?, п?сля того, як пом?тив типов? ознаки у 5-р?чно? д?вчинки.
[11]
[12]
Ген, пов'язаний ?з захворюванням, був уперше вид?лений
Франчезко Рам?резом
у Медичному центр? Маунт Синай (Mount Sinai Medical Center), м?ста Нью-Йорк у 1991.
[13]
Еп?дем?олог?чн? особливост?
[
ред.
|
ред. код
]
Досл?джено, що на синдром Марфана ж?нки ? чолов?ки хвор?ють однаково часто,
[14]
? мутац?я не залежить в?д етн?чних ? географ?чних б?ом?в.
[9]
Частота хворих становить 1:3.000-10.000.
[7]
[9]
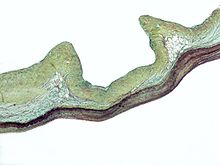 М?кропрепарат, де ? м?ксоматозна дегенерац?я аортального клапану, який часто зустр?ча?ться при синдром? Марфана
М?кропрепарат, де ? м?ксоматозна дегенерац?я аортального клапану, який часто зустр?ча?ться при синдром? Марфана
Синдром Марфана спричиня?ться мутац?ями FBN1 гена 15 хромосоми,
[15]
що коду? ф?брил?н-1 ? гл?копроте?новий компонент екстрацелюлярного матриксу. Ф?брил?н-1 ? необх?дний для правильного формування екстрацелюлярного матриксу, а саме б?огенезу ? п?дтримання еластичних волокон. Екстрацелюлярний матрикс ? критично необх?дний для структурно? ?нтеграц?? сполучно? тканини, а також слугу? резервуаром для фактор?в росту.
[16]
Еластичн? волокна ?снують у кожному куточку людського т?ла, проте найважлив?шу функц?ю виконують в аорт?, сухожилках ? в?йчастому пояску ока; тому ц? зони ? ? найб?льш вразлив?.
Трансгенетичн? миш?-нос?? ?дино? коп?? мутантного ф?брил?ну-1, мутац?? схожо? на ту, яка виника? в людському ген?, спричиню? у мишей розвиток синдрому Марфана. Миш? з цим дефектним геном мають схож? прояви з людськими. Окр?м цього, зниження вм?сту нормального ф?брил?ну-1 породжу? Марфан-пов'язан? захворювання у мишей.
Трансформуючий фактор росту бета (
TGF-β
) в?д?гра? важливу роль у розвитку синдрому Марфана. Ф?брил?н-1 безпосередньо пов'язаний з неактивною формою TGF-β, що збер?га? його неактивн?сть результуючи у в?дсутност? б?олог?чно? активност? останнього. Найпрост?ша модель синдрому Марфана вигляда? приблизно так: зниження р?вня ф?брил?ну-1 спричиню? п?двищення р?вня TGF-β через зворотн?й негативний зв'язок синтезу останнього з зв'язуванням з рецепторами. Хоча не ? доведено вза?мозв'язок TGF-β ? специф?чного патогенезу цього захворювання, проте доведено виникнення запально? реакц?? з вив?льненням базоф?льних протеаз, що власне ? спричиня? пов?льну деградац?ю еластичних волокон та ?нших компонент?в екстрацелюлярного матриксу. Важлив?сть TGF-β у формування цього патолог?чного механ?зму була доведена п?сля в?дкриття синдрому Лу?за-Д?ца, який спричинено зм?нами TGFβR2 гена 3 хромосоми, що коду? рецепторний проте?н до TGF-β.
[17]
Синдром Марфана часто плута?ться з синдромом Лу?за-Д?ца, через практично однаковий кл?н?чний переб?г обох патолог?й.
[18]
 Деформац?я груднини, pectus excavatum, у хворого на синдром Марфана
Деформац?я груднини, pectus excavatum, у хворого на синдром Марфана
Сьогодн? в?домо б?льше 30 р?зних ознак та симптом?в, за допомогою яких можна д?агностувати синдром Марфана. Найб?льш виразн? з них асоц?йован? з к?стковою, серцево-судинною та зоровою системами, проте часто бува? уражена вся сполучна тканина.
Б?льш?сть видимих ознак пов'язан? з к?стковою системою. Багато хворих на синдром Марфана ростом вище середнього зр?ст, мають легку непропорц?йн?сть, довг?, худ? к?нц?вки, тонк? зап'ястя з довгими пальцями ? ступнями (арахнодактил?я). Окр?м цього, хвор? на синдром Марфана можуть мати скол?оз, грудний лордоз, деформовану груднину, високу гнучк?сть суглоб?в, високо розм?щену п?днеб?нну к?стку з деформованим зубним рядом та порушенням прикусу, плоскостоп?сть, сутул?сть ? розтяжки на шк?р?. Ц? порушення можуть спричиняти б?ль в суглобах, к?стках ? м'язах. Деяк? хвор? на синдром Марфана мають порушення мовлення, яке спричинюють вади розвитку - високе п?днеб?ння ? мал? щелепи. Часто у хворих виника? остеоартрит. ?нш? ознаки включають обмежений рух стегон, через атипове розм?щення гол?вки стегново? к?стки в кульшовому суглоб?
[19]
[20]
 Дислокац?я кришталика, при синдром? Марфана, з його ниркопод?бною формою ? розм?щенням б?ля цил?арного т?ла.
Дислокац?я кришталика, при синдром? Марфана, з його ниркопод?бною формою ? розм?щенням б?ля цил?арного т?ла.
При синдром? Марфана порушення зорово? системи р?зноман?тн?, проте в б?льшост? зустр?ча?ться дислокац?я кришталика.
[20]
Це трапля?ться через слабк?сть в?йчастого пояска (цинново? зв'язки) ? сполучнотканинних тяж?в, як? вза?морозташовують кришталик в оц?. В?йчастий поясок м?стить у велик?й к?лькост? ф?брил?н-1, що ? створю? п?двищену пластичн?сть пояска при порушеннях конформац?? б?лка. Нижн? шари цинново? зв'язки мають найб?льш виражену дисфункц?ю, що власне ? спричиня? рух кришталика вгору ? назовн?, у б?льшост? хворих. При синдром? Марфана супутн?ми ? близькозор?сть, проте трапля?ться ? далекозор?сть, особливо у випадках дислокац?? кришталика всередину по в?дношенню до передньо? камери ока. Сублюксац?я (часткова дислокац?я) кришталика визнача?ться кл?н?чно у 80 % пац??нт?в за допомогою сл?т-ламп б?ом?кроскопа.
?нш? ознаки та симптоми пов'язан? з враженнями системи зору включають в себе зб?льшення кривини очного яблука, м?оп?ю, страб?зм, ексотроп?ю та езотроп?ю.
[20]
Серцево-судинна система
[
ред.
|
ред. код
]
Житт?во загрозлив? ознаки ? симптоми, пов'язан? з порушеннями серцево-судинно? системи, ?: слаб?сть, задишка,
тах?кард?я
(пришвидшене серцебиття), загруднинн?й б?ль, який ?нколи ?ррад?ю? в спину, плече або руку. При порушеннях периферично? гемодинам?ки у хворих синдромом Марфана може бути виражена г?потерм?я к?нц?вок. Порушення серцевих тон?в, пров?дно? системи серця, а також симптоматика серцево? ?шем?? повинн? бути сигналом для л?каря до наступних обстежень серцево-судинно? системи. Для хворих на синдром Марфана ? типовим дегенерац?я мед?? аортального та м?трального клапан?в, що спричиня? недостатн?сть (регург?тац?ю) останн?х. Однак найважлив?шою ознакою для д?агностики синдрому Марфана залиша?ться
аневризма
(розширення) аорти. В?дом? випадки безсимптомного переб?гу аневризми аорти, яка п?сля повно? дегенерац?? мед?? розшаровувалась, що власне ? спричиня? п?двищену смертн?сть хворих.
Через порушення сполучно? тканини ?сну? п?двищений ризик дислокац?? м?трального протезу.
[21]
Через це нада?ться перевага х?рург?чному ремоделюванню над протезуванням клапана. У випадку протезування аорти рекомендуються клапан-збережувальн? операц?? (операц?я Дев?да тощо).
Легенева симптоматика не ? головною особлив?стю переб?гу синдрому Марфана,
[22]
проте деколи може виникати спонтанний пневмоторакс.
[23]
П?д час спонтанного одностороннього пневмотораксу, пов?тря потрапля? у плевральний прост?р м?ж грудною кл?ткою ? легенями. Через це виника? компрес?я ? нав?ть колапс легень. Це супроводжу?ться в?дчуттям хворого болю, задишки, ц?анозу ? може спричинити смерть. ?нш? легенев? прояви синдрому Марфана включають н?чне апное
[24]
та ?д?опатичн? обструктивн? легенев? захворювання.
[25]
Патолог?чн? зм?ни у легенях включають кистозн? зм?ни, емф?зему, пневмон??, бронх?ектаз, верх?вковий ф?броз ? вроджен? зм?ни, так? як г?поплаз?я середньо? частки.
[22]
Дуральна ектаз?я ? ослаблення сполучно? тканини
твердо? оболони
спинного мозку часто ? причиною зниження якост? життя хворого. Це ускладнення часто ? нед?агностоване, внасл?док в?дсутност? явних ознак. Симптоматика наступна: б?ль у нижн?й частин? спини, ногах, у живот?, ?нш? невролог?чн? симптоми у нижн?х к?нц?вках та нав?ть головний б?ль. Ц? симптоми б?льш виражен? при горизонтальному положенн? хворого. Дуральна ектаз?я в ранн?х стад?ях розвитку р?дко д?агносту?ться за допомогою рентген-мон?торингу. При магн?тно-резонансному обстеженн? проявля?ться розширеною твердою оболоною поперекових хребц?в.
[26]
?нш? хребцев? проблеми, пов'язан? з синдромом Марфана, включають дегенерац?ю хребцевих диск?в, сп?нальн? кисти ? дисфункц?ю автономно? нервово? системи.
Кожний з батьк?в ма? 50 % передач? генетичного дефекту кожному з д?тей через автосомно-дом?нантну природу мутац??. Б?льш?сть хворих на синдром Марфана мають родича з цим захворюванням. У близько 15-30 % ус?х хворих дана мутац?я виникла вперше;
[16]
так? спонтанн? мутац?? виникають в 1 з 20.000. Синдром Марфана ? також прикладом негативно? дом?нантно? мутац?? ? гаплонедостатност?.
[27]
[28]
Ця мутац?я ма? вар?абельну експресивн?сть, неповна пенентрантн?сть не була задокументована.
Ультразвукове обстеження хворого на синдром Марфана, виявля?ться розширений кор?нь аорти
Д?агностичн? критер?? синдрому Марфана були м?жнародно затвердженими у 1996.
[29]
Д?агноз синдрому Марфана базу?ться на основ? родинно? ?стор?? ? по?днання основних та другорядних ?ндикатор?в порушення, що р?дко зустр?ча?ться в б?льшост? популяц??, наприклад: чотири ознаки порушень к?стково? системи з одн??ю або б?льше ознаками ?нших систем орган?зму, таких як зорового анал?затора чи серцево-судинно?, як? одночасно можна д?агностувати у одного хворого. Наступн? стани можуть виникати внасл?док синдрому Марфана, проте зустр?чаються у людей без цього синдрому.
- Колобома очно? райдужки
[34]
- Вищий середнього р?ст
- Тах?кард?я
[35]
- Кили
- Високо розм?щена п?днеб?нна к?стка
- П?двищена рухлив?сть суглоб?в
- К?фоз
- Недостатн?сть клапан?в серця
- Мальоклюз?я
- М?крогнат?я (мала нижня щелепа)
[34]
- Пролапс м?трального клапана
- М?оп?я (близькозор?сть)
Рев?з?я Гентсько? нозолог?чно? класиф?кац??
[
ред.
|
ред. код
]
 Ознаки долон?: згори ? нормальна долоня, нижче ? долоня хворого на синдром Марфана
Ознаки долон?: згори ? нормальна долоня, нижче ? долоня хворого на синдром Марфана
У 2010 нозолог?я Гента була переглянута ? нов? д?агностичн? критер?? додалися до попередньо? домовленост? 1996. 7 нових критер?й можуть бути використан? для д?агностики:
[40]
[41]
У випадку в?дсутност? родинно? ?стор?? синдрому Марфана:
- Z-score кореня аорти ≥ 2 ? вивих кришталика
- Z-score кореня аорти≥ 2 ? мутац?я FBN1 гена
- Z-score кореня аорти ≥ 2 ? системна оц?нка* > 7 points
- Вивих кришталика ? мутац?я FBN1 з д?агностованою вадою аорти
У випадку наявност? синдрому Марфана в родич?в:
- Вивих кришталика
- Системна оц?нка* ≥ 7
- Z-score кореня аорти≥ 2
- Системна оц?нка:
- Зап'ясткова ? долонна ознака = 3 (у випадку наявност? лише одн??? = 1)
- Pectus carinatum формац?я = 2 (pectus excavatum або асиметр?я грудно? кл?тки = 1)
- Плоскостоп?сть = 2
- Дуральна ектаз?я = 2
- Protrusio acetabuli = 2
- Пневмоторакс= 2
- Зменшення сп?вв?дношення верхньо? ? нижньо? частини т?ла ? зб?льшення сп?вв?дношення довжини рук до висоти ? тяжкий скол?оз = 1
- Скол?оз
або к?фоз = 1
- Знижена рухлив?сть л?ктя = 1
- Лицев? ознаки (3/5) = 1 (дол?хосефал?я, енофтальмоз, звисаюч? пов?ки, г?поплаз?я моляр?в, ретрогнат?я)
- Розтяжки шк?ри (стр??) = 1
- М?оп?я
> 3 diopters = 1
- Пролапс м?трального задньо? стулки м?трального клапана 1?4 1
Долонна ознака (ознака Стайнберга) визнача?ться, якщо у пац??нта г?пермоб?льний л?ктьово-зап'ястний ? променево-зап'ястний суглоби.
Зап'ясткова ознака (ознака Волкера) позитивна, якщо пац??нт може обхопити одн??ю рукою другу, торкаючись великим пальцем м?зинця.
Диференц?йна д?агностика
[
ред.
|
ред. код
]
Багато р?зних порушень можуть продемонструвати симптоматику синдрому Марфана.
[42]
Генетичн? досл?дження ? оц?нка р?зних ознак та симптом?в дозволяють ?х в?др?знити. Наступн? захворювання схож? за переб?гом на синдром Марфана:
Нема? причинного л?кування синдрому Марфана, проте впродовж останн?х тривал?сть життя хворих значно покращилось ? зараз схоже до життя звичайно? людини.
[44]
Синдром л?ку?ться симптоматично, по в?дношенню до ушкоджень р?зних орган?в. У д?тей медикаментозно запоб?га?ться дилатац?я (розширення) аорти.
Регулярний огляд у кард?олога ? необх?дним для спостереження серцевих клапан?в ? аорти. Головною ц?ллю у л?куванн? ? спов?льнення прогрес?? дилатац?? аорти ? ушкодження серцевих клапан?в, за допомогою проф?лактики порушень пров?дно? системи серця (аритм??, тах?кард?я) ? зменшення системного кров'яного тиску.
Медикаментозна терап?я
[
ред.
|
ред. код
]
Медикаментозна терап?я часто включа? бета-блокатори, так? як пропранолол, або у раз? нетолерантност? хворого до нього, блокатор?в кальц??вих канал?в та ACE ?нг?б?тор?в.
[7]
[8]
Через те, що антагон?сти анг?отензин-2 рецептор?в також знижують вм?ст TGF-β. ц? препарати досл?джувались на пац??нтах з тяжким переб?гом синдрому Марфана ? спричинило спов?льнення розширення аорти.
[45]
Проте, нещодавн? досл?дження показали схож? результати п?сля використання ARB, лозартану ? сучасно? бета-блокаторно? терап??, наприклад препарату атенолол.
[46]
Ф?зична активн?сть
[
ред.
|
ред. код
]
Американська Асоц?ац?я Серця (The American Heart Association) ма? наступн? рекомендац?? для хворих синдромом Марфана з в?дсутньою або малою дилятац??ю аорти:
- Скор?ше дозволен? заняття: боул?нг, гольф, сноркл?нг, спортивне ход?ння, тредм?л.
- Середн?й ризик несуть наступн?: баскетбол, рокетбол, сквош, б?г, катання на лижах, футбол, тен?с, бейсбол, катання на мотоцикл?, к?нний спорт.
- Високий ризик несуть наступн?: бод?б?лдинг, вайтл?фтинг, хокей, скелелаз?ння, в?ндсерф?нг, серф?нг, скубадайв?нг.
Х?рург?чн? втручання
[
ред.
|
ред. код
]
Якщо дилятац?я аорти прогресу?, викликаючи розриву або травми кореня аорти, а також призводить до тяжко? недостатност? аортального або ?нших клапан?в, тод? х?рург?чне втручання ста? необх?дним. Протезування аорти це складна операц?я, проте виконана планово ма? значно б?льше шанс?в на усп?х. Саме втручання залежить в?д тяжкост? стану пац??нта ? зараз ?снують операц?? з? збереженням клапанного апарату.
[47]
?сну? тенденц?я ? до зб?льшення к?лькост? ?нших судинних операц?й у хворих на синдром Марфана, через зб?льшення тривалост? життя останн?х, наприклад: протезування низх?дно? частини грудно? аорти а також ?нших судин.
К?стков? ? зоров? прояви синдрому Марфана також можуть бути складними, проте н?коли житт?во небезпечними. Ц? симптоми зазвичай л?куються типово, наприклад р?зними знеболювальними або м'язовими релаксантами. Через те, що синдром Марфана може спричинити асимптоматичн? спинн? деформац??, р?зного роду хребтов? операц?? мусять виконуватись з особливою обережн?стю, незважаючи на складн?сть само? операц??.
Л?кування спонтанного пневмотораксу залежить в?д к?лькост? пов?тря у плевральному простор? ? переб?гу ускладнення у конкретного хворого. Невеликий пневмоторакс може самовир?шитись без активного втручання протягом 1-2 тижн?в. Рецидивуюч? пневмоторакси можуть потребувати х?рург?чного втручання. Тяжк? пневмоторакси вимагають грудно? дренажно? системи, яку використовують протягом дек?лькох дн?в. Велик? пневмоторакси ? ургентними станами, як? вимагають ургентно? декомпрес??.
Упродовж ваг?тност?, нав?ть за в?дсутност? серцево-судинних аномал?й, ж?нки з синдромом Марфана мають високий ризик розшарування аорти, яке часто ? фатальним за в?дсутност? негайного х?рург?чного втручання. Ж?нки з синдромом Марфана повинн? проходити ехокард?ограф?чне обстеження кожн? 6-10 тижн?в упродовж ваг?тност?, для оц?нки д?аметра кореня аорти. П?д час пер?оду полог?в б?льшост? викону?ться кесар?в розтин.
[48]
Синдром Марфана експресу?ться дом?нантно. Це означа?, що дитина одного з батьк?в, який переносить мутований ген, ма? 50 % шанс народитись хворою. У 1996 роц? був виконаний перший перед?мплантац?йний генетичний тест.
[49]
Це означа?, що на ранньому етап? ваг?тност? було проведене генетичне тестування, для визначення ? переривання ваг?тност? у випадку наявност? в ембр?она мутованого гена, в?дпов?дального за синтез ф?брил?на-1.
Завдяки сучасн?й серцевосудинн?й х?рург?? ? терап?? лозартаном ? метопрололом, прогноз у хворих на синдром Марфана позитивний. Проте, ран?ше тривал?сть життя у вищезгаданих була знижена в середньому на третину. Б?льш?сть помирали у п?дл?тковому в?ц? через серцево-судинн? проблеми. Сьогодн? адекватне проф?лактичне л?кування ? рання дозволя? уникнути б?льшост? симптом?в. Отже ?сну? тенденц?я до подовження життя хворих на синдром Марфана.
[50]
В?домо що ж?нки з Марфаном в середньому живуть довше.
Сусп?льство ? культура
[
ред.
|
ред. код
]
Популяризатори синдрому Марфана включають:
- Фло Г?мен (Flo Hyman), ол?мп?йська ср?бна медал?стка з ж?ночого волейболу (1984), яка раптово померла п?д час матчу в?д розшарування аорти.
[51]
- Джонатан Ларсон (Jonathan Larson), автор ? композитор Рент (
Rent)
, який помер в?д розшарування аорти, день перед музичним виступом.
[52]
[53]
- В?нсент Ш?авелл? (Vincent Schiavelli), актор ? представницьке лице Фундац?? Марфана (The Marfan Foundation), п?зн?ше перейменована в Нац?ональну Фундац?ю Марфана, який хвор?в на синдром Марфана, але помер в?д нев?домо? причини.
- Музикант Бредфорд Кокс (Bradford Cox) з ?нд?-рок гурту Д?ргантер (Deerhunter).
[54]
- Офшор Еван Робертсон (Ewan Robertson), музикант ? граф?чний редактор Б?уг Дада Рекорд?нгс (Big Dada Recordings), який раптово помер п?д час серцево? операц??, пов'язано? з синдромом Марфана.
[55]
- ?саах Овст?н (Isaiah Austin), гравець баскетбольно? команди, д?агностований з синдромом Марфана, через що мус?в покинути кар'?ру NBA(Ен-Б?-Ей).
[56]
[57]
- Жав??р Ботет (Javier Botet), ?спанський актор, який грав рол? надприродних створ?нь у ф?льмах жах?в, таких як: REC,
Mama
[58]
? The Strain телев?з?йн? сер??.
[59]
Досл?дники вважають, що Ахенатен (
Akhenaten
), фараон ?гипту 18 покол?ння м?г хвор?ти синдромом Марфана.
[60]
[61]
Авраам Л?нкольн (
Abraham Lincoln)
вважався протягом певного часу хворим на синдром Марфана,
[62]
проте ця точка зору була спростована сучасними генетиками
[63]
[64]
. Отримання точного результату ? неможливим, через в?дмову отримання зразка ДНК.
[65]
- ↑
а
б
http://www.marfan.org/about/signs
- ↑
https://my.clevelandclinic.org/health/diseases/17209-marfan-syndrome/symptoms
- ↑
а
б
в
г
д
https://www.nhlbi.nih.gov/health-topics/marfan-syndrome
- ↑
http://omim.org/entry/154700
- ↑
а
б
в
г
д
е
ж
What Is Marfan Syndrome?
.
NHLBI, NIH
. 1 жовтня 2010. Арх?в
ориг?налу
за 6 травня 2016
. Процитовано 16 травня 2016
.
- ↑
What Are the Signs and Symptoms of Marfan Syndrome?
.
NHLBI, NIH
. 1 жовтня 2010. Арх?в
ориг?налу
за 11 червня 2016
. Процитовано 16 травня 2016
.
- ↑
а
б
в
г
д
е
Marfan Syndrome
.
National Organization for Rare Disorders
. 2014. Арх?в
ориг?налу
за 12 листопада 2019
. Процитовано 16 травня 2016
.
- ↑
а
б
в
How Is Marfan Syndrome Treated?
.
NHLBI, NIH
. 1 жовтня 2010. Арх?в
ориг?налу
за 11 червня 2016
. Процитовано 16 травня 2016
.
- ↑
а
б
в
г
Medical management of Marfan syndrome
.
Circulation
. Т. 117, № 21. 2008. с. 2802?13.
doi
:
10.1161/CIRCULATIONAHA.107.693523
.
PMID
18506019
. Арх?в
ориг?налу
за 31 с?чня 2010
. Процитовано 27 вересня 2016
.
- ↑
Marfan, Antoine (1896). Un cas de deformation congenitale des quartre membres, plus prononcee aux extremities, caracterisee par l'allongement des os avec un certain degre d'amincissement [A case of congenital deformation of the four limbs, more pronounced at the extremities, characterized by elongation of the bones with some degree of thinning].
Bulletins et Memoires de la Societe Medicale des Hopitaux de Paris
(фр.)
.
13
(3rd series): 220?226.
OCLC
493643386
.
Шаблон:NAID
.
- ↑
Antoine Bernard-Jean Marfan
.
Whonamedit?
.
Арх?в
ориг?налу за 8 March 2016
. Процитовано 16 May 2016
.
- ↑
Johns Hopkins Comprehensive Marfan Center
. Арх?в
ориг?налу
за 15 жовтня 2008
. Процитовано 27 вересня 2016
.
- ↑
Brown P (July 27, 1991).
- ↑
Determinants of quality of life in Marfan syndrome
.
Psychosomatics
. Т. 49, № 3. 2008. с. 243?8.
doi
:
10.1176/appi.psy.49.3.243
.
PMID
18448780
. Арх?в
ориг?налу
за 13 липня 2012
. Процитовано 27 вересня 2016
.
- ↑
McKusick V (1991). The defect in Marfan syndrome.
Nature
. Т. 352, № 6333. с. 279?81.
Bibcode
:
1991Natur.352..279M
.
doi
:
10.1038/352279a0
.
PMID
1852198
.
- ↑
а
б
Cotran; Kumar, Collins (1998).
Robbins Pathologic Basis of Disease
. Philadelphia: W.B Saunders Company.
ISBN
0-7216-7335-X
.
- ↑
Entrez Gene (2007).
TGFBR2 transforming growth factor, beta receptor II
. NCBI. Арх?в
ориг?налу
(Entrez gene entry)
за 29 травня 2020
. Процитовано 11 с?чня 2007
.
- ↑
Related Disorders: Loeys-Dietz
. National Marfan Foundation. Арх?в
ориг?налу
за 25 вересня 2006
. Процитовано 11 с?чня 2007
.
- ↑
Van de Velde, S; Fillman, R; Yandow, S (2006). Protrusio acetabuli in Marfan syndrome. History, diagnosis, and treatment.
The Journal of bone and joint surgery. American volume
. Т. 88, № 3. с. 639?46.
doi
:
10.2106/JBJS.E.00567
.
PMID
16510833
.
- ↑
а
б
в
OMIM Entry - # 154700 - MARFAN SYNDROME; MFS
.
omim.org
. Арх?в
ориг?налу
за 8 вересня 2019
. Процитовано 8 серпня 2016
.
- ↑
Zipes, Libby Bonow Braunwald (2005).
Braunwald's Heart Disease ~ A Textbook of Cardiovascular Medicine, Seventh Edition
. United States of America: Elseview Saunders. с. 1894.
ISBN
0-7216-0509-5
.
- ↑
а
б
Dyhdalo, K; Farver, C (2011).
Pulmonary histologic changes in Marfan syndrome: a case series and literature review
.
American journal of clinical pathology
. Т. 136, № 6. с. 857?63.
doi
:
10.1309/AJCP79SNDHGKQFIN
.
PMID
22095370
.
- ↑
Siepe, M; Loffelbein, F (2009). [The Marfan syndrome and related connective tissue disorders].
Medizinische Monatsschrift fur Pharmazeuten
. Т. 32, № 6. с. 213?9.
PMID
19554831
.
- ↑
а
б
Kohler, M.; Blair, E.; Risby, P.; Nickol, A. H.; Wordsworth, P.; Forfar, C.; Stradling, J. R. (1 лютого 2009).
The prevalence of obstructive sleep apnoea and its association with aortic dilatation in Marfan's syndrome
.
Thorax
. Т. 64, № 2. с. 162?166.
doi
:
10.1136/thx.2008.102756
.
ISSN
1468-3296
.
PMID
18852161
. Арх?в
ориг?налу
за 13 травня 2016
. Процитовано 27 вересня 2016
.
- ↑
Corsico, A. G.; Grosso, A.; Tripon, B.; Albicini, F.; Gini, E.; Mazzetta, A.; Di Vincenzo, E. M.; Agnesi, M. E.; Tsana Tegomo, E. (1 червня 2014).
Pulmonary involvement in patients with Marfan Syndrome
.
Panminerva Medica
. Т. 56, № 2. с. 177?182.
ISSN
1827-1898
.
PMID
24994580
. Арх?в
ориг?налу
за 14 травня 2016
. Процитовано 27 вересня 2016
.
- ↑
Marfan Syndrome
. Mayo Clinic. Арх?в
ориг?налу
за 10 с?чня 2007
. Процитовано 12 с?чня 2007
.
- ↑
Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome
.
The Journal of Clinical Investigation
. Т. 114, № 2. 2004. с. 172?81.
doi
:
10.1172/JCI20641
.
PMC
449744
.
PMID
15254584
.
- ↑
Marfan's syndrome.
Lancet
. Т. 366, № 9501. 2005. с. 1965?76.
doi
:
10.1016/S0140-6736(05)67789-6
.
PMC
1513064
.
PMID
16325700
.
- ↑
Revised diagnostic criteria for the Marfan syndrome.
Am. J. Med. Genet
. Т. 62, № 4. 1996. с. 417?26.
doi
:
10.1002/(SICI)1096-8628(19960424)62:4<417::AID-AJMG15>3.0.CO;2-R
.
PMID
8723076
.
- ↑
Marfan syndrome. Long-term survival and complications after aortic aneurysm repair
.
Circulation
. Т. 91, № 3. 1995. с. 728?33.
doi
:
10.1161/01.CIR.91.3.728
.
PMID
7828300
. Арх?в
ориг?налу
за 8 червня 2011
. Процитовано 27 вересня 2016
.
- ↑
Marfan Syndrome: Signs and Symptoms
. www.ucsfhealth.org. Арх?в
ориг?налу
за 17 червня 2010
. Процитовано 28 серпня 2009
.
- ↑
What is Marfan Syndrome?
. Marfan Trust. Арх?в
ориг?налу
за 10 червня 2015
. Процитовано 1 червня 2015
.
- ↑
Marfan Syndrome: The Similarities to Copper Deficiency
. www.ctds.info. Арх?в
ориг?налу
за 21 лютого 2009
. Процитовано 29 серпня 2009
.
- ↑
а
б
в
MedlinePlus Encyclopedia
Marfan syndrome
- ↑
Marfan syndrome
.
Genetics Home Reference
. U.S. National Institute of Health. Арх?в
ориг?налу
за 29 серпня 2009
. Процитовано 28 серпня 2009
.
- ↑
The bone mineral status of patients with Marfan syndrome.
Journal of Bone and Mineral Research
. Т. 10, № 10. 1995. с. 1550?5.
doi
:
10.1002/jbmr.5650101017
.
PMID
8686512
.
- ↑
Northwestern Memorial Center for Heart Valve Disease.
- ↑
а
б
About Marfan Syndrome: Features
. National Marfan Foundation. Арх?в
ориг?налу
за 20 серпня 2009
. Процитовано 28 серпня 2009
.
- ↑
Living with Marfan Syndrome: Dental issues
. National Marfan Foundation. Арх?в
ориг?налу
за 6 вересня 2009
. Процитовано 28 серпня 2009
.
- ↑
2010 Revised Ghent Nosology
. National Marfan Foundation. Арх?в
ориг?налу
за 14 с?чня 2011
. Процитовано 31 с?чня 2011
.
- ↑
Loeys, BL; Dietz, HC; Braverman, AC; Callewaert, BL; De Backer, J; Devereux, RB; Hilhorst?Hofstee, Y; Jondeau, G; Faivre, L (2010).
The revised Ghent nosology for the Marfan syndrome
(PDF)
.
Journal of Medical Genetics
. Т. 47, № 7. London: BMJ Group. с. 476?485.
doi
:
10.1136/jmg.2009.072785
.
ISSN
0022-2593
.
OCLC
857424767
.
PMID
20591885
.
Арх?в
(PDF)
ориг?налу за 10 с?чня 2016
. Процитовано 27 вересня 2016
.
- ↑
Emery and RImoin's Principles and Practice of Medical Genetics. 5th ed
. Philadelphia, Pennsylvania: Churchill Livingstone Elsevier. 2007.
- ↑
Greally та GeneReviews, 2010
- ↑
Questions and Answers about Marfan Syndrome
. Niams.nih.gov. Арх?в
ориг?налу
за 9 кв?тня 2014
. Процитовано 23 червня 2014
.
- ↑
Pyeritz RE (2008). A small molecule for a large disease.
NEJM
. Т. 358, № 26. с. 2829?31.
doi
:
10.1056/NEJMe0804008
.
PMID
18579819
.
- ↑
R.V. Lacro et al.:
Atenolol versus Losartan in Children and Young Adults with Marfan's Syndrome
.
- ↑
Heart Surgery for Marfan Syndrome
. Mayo Clinic. Арх?в
ориг?налу
за 18 грудня 2006
. Процитовано 12 с?чня 2007
.
- ↑
Chen H (2007).
Marfan Syndrome
. eMedicine. Арх?в
ориг?налу
за 6 липня 2009
. Процитовано 25 червня 2007
.
- ↑
Preimplantation genetic testing for Marfan syndrome
.
Mol. Hum. Reprod
. Т. 2, № 9. 1996. с. 713?5.
doi
:
10.1093/molehr/2.9.713
.
PMID
9239687
.
- ↑
Keane, Martin G.; Pyeritz, Reed E. (2008).
Medical Management of Marfan Syndrome
.
Circulation
. Т. 117, № 21. с. 2802?2813.
doi
:
10.1161/CIRCULATIONAHA.107.693523
.
ISSN
1524-4539
.
PMID
18506019
. Арх?в
ориг?налу
за 20 березня 2016
. Процитовано 27 вересня 2016
.
- ↑
Flo Hyman
. Volleyball Hall of Fame. Арх?в
ориг?налу
за 30 с?чня 2008
. Процитовано 6 с?чня 2009
.
- ↑
Lawrence Van Gelder (13 грудня 1996).
On the Eve of a New Life, an Untimely Death
.
The New York Times
. Арх?в
ориг?налу
за 14 травня 2020
. Процитовано 17 липня 2008
.
- ↑
Kirk, Fiona J. (26 липня 2011).
Syndrome survival: New drugs offer promise for often-fatal Marfan tissue disorder
.
The Daily
. Арх?в
ориг?налу
за 17 травня 2020
. Процитовано 24 листопада 2011
.
- ↑
Deerhunter interview
.
Pitchfork.com
. 11 червня 2007. Арх?в
ориг?налу
за 6 жовтня 2014
. Процитовано 1 жовтня 2014
.
(Interviewer): i think a lot of people still don't know [that you have Marfan Syndrome]... (Bradford Cox): People think I'm a junkie.
- ↑
Big Dada to release final, posthumous album by Offshore
. FACT Magazine. 14 липня 2015. Арх?в
ориг?налу
за 22 кв?тня 2016
. Процитовано 13 серпня 2016
.
- ↑
NBA makes Austin's dreams come true with gesture at draft
.
Houston Chronicle
. 26 червня 2014. Арх?в
ориг?налу
за 26 серпня 2018
. Процитовано 27 вересня 2016
.
- ↑
Isaiah Austin has Marfan syndrome
.
ESPN.com
. 22 червня 2014. Арх?в
ориг?налу
за 22 червня 2014
. Процитовано 22 червня 2014
.
- ↑
Chang, Justin (15 с?чня 2013).
Mama
.
Variety
. Арх?в
ориг?налу
за 25 грудня 2014
. Процитовано 27 вересня 2016
.
- ↑
The Creature and Makeup Effects of The Strain - Tested.com
.
Tested
. Арх?в
ориг?налу
за 18 вересня 2016
. Процитовано 9 лютого 2016
.
- ↑
The Mystery of Akhenaten: Genetics or Aesthetics?
. Арх?в
ориг?налу
за 8 лютого 2010
. Процитовано 27 вересня 2016
.
- ↑
Akhenaten's illness
. Арх?в
ориг?налу
за 26 с?чня 2014
. Процитовано 27 вересня 2016
.
- ↑
Marion R (1994). Mr. Lincoln and Dr. Marfan's syndrome.
Was George Washington Really the Father of Our Country?
. Reading, MA: Addison-Wesley.
- ↑
Sotos, John G.
The Physical Lincoln: Other Medical Theories
.
www.physical-lincoln.com
. Mt. Vernon Book Systems. Арх?в
ориг?налу
за 9 серпня 2016
. Процитовано 13 серпня 2016
.
Medical Condition: Marfan syndrome; Did Lincoln Have It? No
- ↑
Sotos JG (2008).
The Physical Lincoln Sourcebook
. Mt. Vernon, VA: Mt. Vernon Book Systems. с. 29.
ISBN
978-0-9818193-3-4
.
- ↑
Ready T (1999). Access to presidential DNA denied.
Nature Medicine
. Т. 5, № 8. с. 859.
doi
:
10.1038/11287
.
PMID
11645164
.