 Порамнува?ето
на целиот
геном
е вообичаен метод во споредбената геномика. Ова порамнува?е на осум геноми на бактери?ата
Yersinia
открива 78 локално колинеарни блокови конзервирани ме?у сите осум
таксони
. Секо? хромозом е поставен хоризонтално и
хомологните
блокови во секо? геном се прикажани како идентично обоени региони поврзани ме?у геномите. Регионите кои се превртени во однос на геномот KIM на
Y. pestis
се поместени под средишната оска на геномот.
[1]
Порамнува?ето
на целиот
геном
е вообичаен метод во споредбената геномика. Ова порамнува?е на осум геноми на бактери?ата
Yersinia
открива 78 локално колинеарни блокови конзервирани ме?у сите осум
таксони
. Секо? хромозом е поставен хоризонтално и
хомологните
блокови во секо? геном се прикажани како идентично обоени региони поврзани ме?у геномите. Регионите кои се превртени во однос на геномот KIM на
Y. pestis
се поместени под средишната оска на геномот.
[1]
Споредбена геномика
― поле на
биолошко истражува?е
во кое се споредувани
геномските
особини на различни
организми
.
[2]
[3]
Геномските особини може да вклучуваат
низа на ДНК
,
гени
, редослед на гени, регулаторни низи и други геномски структурни обележ?а.
[3]
Во оваа гранка на
геномиката
, цели или големи делови од геномите кои произлегуваат од геномските проекти, се споредувани за проучува?е на основните биолошки сличности и разлики, како и
еволутивните
односи ме?у организмите.
[2]
[4]
[5]
Главното начело на споредбената геномика е дека заедничките особини на два организми честопати ?е бидат кодирани во
ДНК
што еволутивно е зачувана ме?у нив.
[6]
Затоа, споредбените геномски пристапи започнуваат со праве?е некаков облик на
усогласува?е
на геномските секвенци и бара?е ортологни секвенци (секвенци кои имаат
заедничко потекло
) во подредените геноми и проверка до ко? степен тие низи се зачувани. Врз основа на нив, заклучувани се геномот и молекуларната еволуци?а и тоа може да биде ставено во контекст на, на пример,
фенотипска
еволуци?а или
популациона генетика
.
[7]
Виртуелно започна веднаш штом целиот геном на два организми станаа достапни (т.е. геномите на бактериите
Haemophilus influenzae
and
Mycoplasma genitalium
) во 1995 година, споредбената геномика сега е стандардна компонента на анализата на секо?а нова геномска низа.
[2]
[8]
Со зголемува?ето на бро?от на проекти за геном поради напредокот во
технологиите
на
секвенционира?е на ДНК
, особено методите за масивното напоредно секвенционира?е кон кра?от на 2000-тите, ова поле станало пософистицирано, што овозможува да се справи со многу геноми во една студи?а.
[9]
Споредбената геномика открила високи нивоа на сличност поме?у тесно поврзани организми, како што се
лу?ето
и
шимпанзата
, и, уште поизненадувачки, сличноста поме?у навидум далечно поврзани организми, како што се лу?ето и
квасецот
Saccharomyces cerevisiae
.
[4]
Исто така, ?а покажа кра?ната разновидност на составот на гените во различни еволутивни лози.
[8]
Поврзано
:
Истори?а на геномиката
Споредбената геномика има корен во споредбата на
геномите
на
вирусите
во раните 1980-ти.
[8]
На пример, малите РНК вируси што инфицираат
животни
(пикорнавируси) и оние што ги инфицираат
растени?ата
(грашест мозаичен вирус) биле споредени и било покажано дека споделуваат знача?на сличност во низата и, делумно, редоследот на нивните
гени
.
[10]
Во 1986 година, била об?авена првата споредбена геномска студи?а во поголем обем, споредува??и ги геномите на вирусот варичела-зостер и Епшта?н-Баровиот вирус кои содржеле пове?е од 100 гени по секо?.
[11]
Првата целосна геномска низа на клеточен организам, онаа на
Haemophilus influenzae
Rd, била об?авена во 1995 година.
[12]
Вториот труд за секвенционира?е на геномот бил за малата
паразитска
бактери?а
Mycoplasma genitalium
об?авен во истата година.
[13]
Поа?а??и од ово? труд, извештаите за новите геноми неизбежно станале споредбено-геномски студии.
[8]
Микробиолошки геноми
Првиот систем за споредба на целиот геном со висока резолуци?а на микробиолошки геноми од 10-15 kbp бил развиен во 1998 година од Арт Делчер, Са?мон Касиф и Стивен Салцберг и применет за споредба на цели високо поврзани
микробни
организми со нивните соработници во Институтот за геномски истражува?а. Системот се нарекува MUMmer и е опишан во публикаци?а во списанието
Nucleic Acids Research
во 1999 година. Системот им помага на истражувачите да идентификуваат големи преуредува?а,
мутации
на единечна основа, превртува?а, тандемско повторувачки шире?а и други полиморфизми. Ка? бактериите, MUMmer овозможува идентификаци?а на полиморфизми кои се одговорни за вирулентност, патогеност и отпорност на
антибиотици
. Системот бил применет и на Проектот за минимален организам при Институтот за геномски истражува?а и последователно на многу други проекти за споредбена геномика.
Геноми на еукариоти
Saccharomyces cerevisiae
, лебниот квасец, бил првиот
еукариот
на ко? неговата целосна геномска низа била об?авена во 1996 година.
[14]
По об?авува?ето на геномот на кружниот црв
Caenorhabditis elegans
во 1998 година
[15]
и заедно со геномот на овошната мува
Drosophila melanogaster
во 2000 година,
[16]
?ералд М. Рубин и неговата група об?авиле труд со наслов ?
Споредбена геномика на еукариотите
“, во ко? тие ги споредиле геномите на еукариотите
D. melanogaster
,
C. elegans
и
S. cerevisiae
, како и
прокариотот
H. influenzae
.
[17]
Во исто време, Бони Бергер, Ерик Ландер и нивната група об?авиле труд за споредба на целиот геном на
човекот
и глушецот.
[18]
Со об?авува?ето на големите геноми на
'рбетниците
во 2000-тите, вклучително и
човекот
, ?апонската надувувачка риба
Takifugu rubripes
и глушецот, претходно пресметаните резултати од споредбите на големиот геном биле об?авени за презема?е или за гледа?е во геномски прелистувач. Наместо да преземаат свои анализи, пове?ето
биолози
можат да пристапат до овие големи споредби ме?у
видовите
и да ?а избегнат непрактичноста предизвикана од големината на геномите.
[19]
Методите за масивно напоредното секвенционира?е, кои првпат биле воведени во 2007 година, произвеле огромна количина на геномски податоци и им овозможиле на истражувачите да создаваат пове?екратни (прокариотски) нацрт-геномски низи одеднаш. Овие методи, исто така, можат брзо да откри?ат полиморфизми, вметнува?а и брише?а на единечен нуклеотид со картира?е на несклопени чита?а против добро забележан референтен геном, и на то? начин да обезбедат список на можни генски разлики што може да бидат основа за какви било функционални вари?ации ме?у соевите.
[9]
Еден особина на
биологи?ата
е еволуци?ата, еволутивната теори?а е и теоретска основа на споредбената геномика, а во исто време резултатите од споредбената геномика невидено ?а збогатиле и ?а развиле теори?ата на еволуци?ата. Кога се споредувани две или пове?е од геномската низа, може да бидат заклучени еволутивните односи на низите во
филогенетското дрво
. Врз основа на различни податоци од биолошкиот
геном
и проучува?е на вертикалните и хоризонталните постапки на еволуци?ата, може да бидат разбрани виталните делови од структурата на
генот
и неговата регулаторна функци?а.
Сличноста на сродните геноми е основата на споредбената геномика. Ако две суштества имаат неодамнешен заеднички предок, разликите поме?у геномите на двата
вида
еволуирале од геномот на предците. Колку е поблиска врската поме?у два
организми
, толку се поголеми сличностите ме?у нивните геноми. Ако постои блиска врска ме?у нив, тогаш нивниот геном ?е прикаже линеарно однесува?е (синтени?а), имено некои или сите генетски низи се зачувани. Така, геномските низи може да бидат користени за да биде идентификувана функци?ата на генот, со анализа на нивната хомологи?а (сличност на низата) со гени со позната функци?а.
 Човечкиот
ген FOXP2 и еволутивното зачувува?е се прикажани во и пове?екратно порамнува?е (на дното на сликата) на оваа слика од UCSC Genome Browser (прелистувач на геноми на Универзитетот во Калифорни?а). Забележете дека зачувува?ето има тежнее?е да биде групирано околу регионите за кодира?е (егзони).
Човечкиот
ген FOXP2 и еволутивното зачувува?е се прикажани во и пове?екратно порамнува?е (на дното на сликата) на оваа слика од UCSC Genome Browser (прелистувач на геноми на Универзитетот во Калифорни?а). Забележете дека зачувува?ето има тежнее?е да биде групирано околу регионите за кодира?е (егзони).
Ортологните низи се сродни низи во различни видови: ген постои во првобитниот вид, видот поделен на два вида, така што гените во новите видови се ортологни на низата во првобитниот вид. Паралогните низи се одво?уваат со генско
клонира?е
(удво?ува?е на гените): ако одреден ген во геномот е копиран, тогаш копи?ата од двете секвенци е паралогна на првобитниот ген. Еден пар ортологни низи се нарекуваат ортологни парови (ортолози), еден пар паралогни низи се нарекувани колатерални парови (паралози). Ортологните парови обично имаат иста или слична функци?а, што не е нужно случа? за колатералните парови. Во колатералните парови, низите имаат тежнее?е да еволуираат во различни функции.
Споредбената геномика ги искористува и сличностите и разликите во
белковините
,
РНК
и регулаторните региони на различни организми за да заклучи како
одбира?ето
де?ствувала врз овие елементи. Оние елементи кои се одговорни за сличностите ме?у различните видови треба да бидат зачувани низ времето (стабилизирачко одбира?е), додека оние елементи одговорни за разликите ме?у видовите треба да бидат дивергентни (позитивно одбира?е). Конечно, оние елементи кои не се важни за еволутивниот успех на организмот нема да бидат зачувани (одбира?ето е неутрално).
Една од важните цели на теренот е идентификаци?а на механизмите на еволуци?ата на
еукариотскиот
геном. Ме?утоа, често е усложувано со мноштвото настани што се случиле низ истори?ата на поединечните лози, остава??и само искривени и надредени траги во геномот на секо? жив организам. Поради оваа причина, споредбените геномски студии на мали
моделни организми
(на пример, моделот
Caenorhabditis elegans
и тесно поврзаниот
Caenorhabditis briggsae
) се од голема важност за да биде унапредено разбира?ето за општите механизми на еволуци?ата.
[20]
[21]
Сметачките пристапи се неопходни за споредува?е на
геномот
, со оглед на големиот бро? на податоци кодирани во геномите. Многу алатки сега се ?авно достапни, почнува??и од споредби на целиот геном до анализа на
генско изразува?е
.
[22]
Ова ги вклучува пристапите од системите и контролата, теори?ата на информации, анализата на соеви и
рударе?ето податоци
.
[23]
Сметачките пристапи ?е останат критични за истражува?е и настава, особено кога
науката
за информации и
биологи?ата
на геномот се изучувани заедно.
[24]
 Филогенетско дрво
на потомци и реконструирани предци. Бо?ата на гранката ги претставува стапките на точки на прекин во RACF (точки на прекин на милион години). Црните гранки претставуваат неодредени стапки на точка на прекин. Боите на врвовите го прикажуваат соседството на склопот: црна, склоп на геном на ниво на скеле; зелена, склоп на геном на ниво на хромозом; жолта, склоп на геном на ниво на скеле во скала на хромозом. Броевите до ими?ата на видовите означуваат диплоиден бро? на хромозом (ако е познат).
[25]
Филогенетско дрво
на потомци и реконструирани предци. Бо?ата на гранката ги претставува стапките на точки на прекин во RACF (точки на прекин на милион години). Црните гранки претставуваат неодредени стапки на точка на прекин. Боите на врвовите го прикажуваат соседството на склопот: црна, склоп на геном на ниво на скеле; зелена, склоп на геном на ниво на хромозом; жолта, склоп на геном на ниво на скеле во скала на хромозом. Броевите до ими?ата на видовите означуваат диплоиден бро? на хромозом (ако е познат).
[25]
Споредбената геномика започнува со основни споредби на големината на геномот и
генската
густина. На пример, големината на геномот е важна за капацитетот за кодира?е и можеби поради регулаторни причини. Високата генска густина ?а олеснува прибелешката на геномот, анализата на селекци?а на животната средина. Спротивно на тоа, ниската генска густина го попречува картира?ето на генетските
болести
како во
човечкиот
геном.
Порамнува?ата
се користени за забележува?е информации за слични низи како што се потеклото, заедничкото
еволутивно
потекло или заедничката структура и функци?а. Порамнува?а може да бидат направени и за
генските
и за
белковинските
низи.
[26]
[27]
Порамнува?ата се состо?ат од месни или општи порамнува?а во пар, и порамнува?а на пове?е низи. Еден начин да бидат на?дени општи порамнува?а е да биде користен динамичен
програмски
алгоритам
познат како
Нидлман-Вуншовиот алгоритам
. Ово? алгоритам може да биде изменет и да биде користен за нао?а?е месни порамнува?а.
 Пример за филогенетско дрво создадено од порамнува?е на 250 уникатни белковински низи на шилести од
семе?ството
Betacoronavirus.
Пример за филогенетско дрво создадено од порамнува?е на 250 уникатни белковински низи на шилести од
семе?ството
Betacoronavirus.
Филогенетска реконструкци?а
[
уреди
|
уреди извор
]
Друг сметачки метод за споредбена
геномика
е филогенетската реконструкци?а. Користена е за опишува?е на
еволутивните
односи во однос на заедничките
предци
. Врските обично се претставени во дрво наречено
филогенетско дрво
. Слично на тоа, теори?ата за спо?ува?е е ретроспективен модел за следе?е на
алели
на
ген
во
население
на единствена копи?а од предците споделена од членовите на населението. Ова е исто така познато како
на?нов заеднички предок
. Анализата заснована на теори?ата на спо?ува?е се обидува да го предвиди времето поме?у воведува?ето на
мутаци?а
и одреден алел или генска распределба во населението. Ово? временски период е еднаков на тоа колку долго постоел на?новиот заеднички предок. Односите на наследува?е се гледани во облик слична на филогенетско дрво. Соединува?ето (или генеалоги?ата на гените) може да биде видено со помош на дендрограми.
[28]
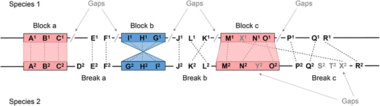 Пример за блокира?е и прекин на синтени?а. Гените сместени на хромозомите од два
вида
се означени со букви. Секо? ген е поврзан со бро? ко? го претставува видот на ко? му припа?а (вид 1 или 2). Ортологните гени се поврзани со испрекинати линии, а гените без ортологна врска се третирани како празнини во програмите за синтени?а.
[29]
Пример за блокира?е и прекин на синтени?а. Гените сместени на хромозомите од два
вида
се означени со букви. Секо? ген е поврзан со бро? ко? го претставува видот на ко? му припа?а (вид 1 или 2). Ортологните гени се поврзани со испрекинати линии, а гените без ортологна врска се третирани како празнини во програмите за синтени?а.
[29]
Дополнителен метод во споредбената геномика е генетското картира?е. Во генетското картира?е, гледа?ето на синтени?ата е еден начин да биде виден зачуваниот редослед на
гени
на
хромозомите
. Обично е користено за хромозоми од сродни
видови
, кои се резултат на заеднички предок.
[30]
Ово? и други методи можат да фрлат светлина врз
еволутивната
истори?а. Една неодамнешна студи?а користела споредбена геномика за реконструкци?а на 16
кариотипови
на предците низ
филогени?ата
на
цицачите
. Сметачката реконструкци?а покажала како хромозомите биле преуредени за време на еволуци?ата на цицачите. Таа дала увид во зачувува?ето на одредени региони кои често се поврзувани со контролата на разво?ните постапки. Покра? тоа, таа помогнала да биде обезбедено разбира?е за еволуци?ата на хромозомите и
генетските болести
поврзани со преуредува?ето на
ДНК
.
 Слика од студи?ата ?Еволуци?а на кариотипот на предците на цицачите и синтенските региони“. Тоа е гледа?е на еволутивната истори?а на реконструирани хромозоми на цицачи врз основа на
човечката
лоза.
[25]
Слика од студи?ата ?Еволуци?а на кариотипот на предците на цицачите и синтенските региони“. Тоа е гледа?е на еволутивната истори?а на реконструирани хромозоми на цицачи врз основа на
човечката
лоза.
[25]
Сметачките алатки за анализа на низи и целосни
геноми
брзо се развивани поради достапноста на голема количина геномски податоци. Во исто време, алатките за споредбена анализа се напредувани и подобрувани. Во предизвиците за овие анализи, многу е важно да бидат видени споредбените резултати.
[31]
Гледа?ето на зачувува?ето на низата е тешка задача на споредбената анализа на низата. Како што е познато, многу е неефикасно рачно да биде испитано порамнува?ето на долгите геномски региони. Прелистувачите за геном кои се на
интернет
обезбедуваат многу корисни алатки за истражува?е на геномските низи поради интегрира?е на сите
биолошки
информации засновани на секвенци за геномските региони. Кога извлекуваме големо количество релевантни биолошки податоци, тие можат да бидат многу лесни за употреба и да одземаат помалку време.
[31]
- UCSC Browser: Ова
мрежно место
?а содржи референтната низа и работните нацрт склопови за голема збирка геноми.
[32]
- Ensembl: Проектот Ensembl произведува геномски бази на податоци за
'рбетници
и други
еукариотски
видови
и ги прави овие информации слободно достапни на интернет.
[33]
- MapView: Прегледувачот на карти обезбедува широк спектар на податоци за картира?е и секвенционира?е на геномот.
[34]
- VISTA е сеопфатен пакет на програми и бази на податоци за споредбена анализа на геномските низи. Изграден е за да бидат видени резултатите од споредбената анализа врз основа на усогласува?ата на ДНК. Претставува?ето на споредбените податоци создадени од VISTA лесно може да одговара и на мали и големи размери на податоци.
[35]
- BlueJay Genome Browser: самосто?на алатка за пове?еразмерно гледа?е на означени геноми и други геномски елементи.
[36]
Предноста на користе?ето семрежни алатки е тоа што овие мрежни места посто?ано се развивани и ажурирани. Има многу нови поставки и содржината може да биде користена семрежно за да биде подобрена ефикасноста.
[31]
Зем?оделството
е област ко?а ги користи придобивките од споредбената
геномика
. Идентификува?ето на
локусите
на поволните
гени
е клучен чекор во одгледува?ето култури кои се оптимизирани за поголем принос, економичност, квалитет и отпорност кон болести. На пример, една студи?а за поврзува?е на ниво на геном, спроведена на 517 домородни сорти
ориз
, открила 80 локуси поврзани со неколку категории на агрономски перформанси, како што се тежината на зрната, содржината на амилоза и толеранци?ата кон суша. Многу од локусите биле претходно некарактеризирани.
[37]
Оваа методологи?а не само што е мо?на, туку е и брза. Претходните методи за идентификаци?а на локуси поврзани со агрономските перформанси барале неколку генерации на внимателно следено
размножува?е
на матичните соеви, напор ко? одзема многу време и е непотребен за споредбени геномички студии.
[38]
Медицинското
поле, исто така, има корист од проучува?ето на споредбената геномика. Во пристапот познат како обратна вакцинаци?а, истражувачите можат да откри?ат кандидатски
антигени
за разво? на
вакцини
преку анализа на
геномот
на патогенот или семе?ството патогени.
[39]
Примената на споредувачки геномски пристап преку анализа на геномите на неколку поврзани патогени може да доведе до разво? на вакцини кои се пове?езаштитни. Група истражувачи употребила таков пристап за да создаде универзална вакцина за стрептокока од групата Б, група
бактерии
одговорни за тешка неонатална инфекци?а.
[40]
Споредбената геномика, исто така, може да биде користена за создава?е специфичност на вакцините против патогени кои се тесно поврзани со
комензалните
микроорганизми
. На пример, истражувачите користеле споредбена геномска анализа на комензалните и патогените соеви на
E. coli
за да ги идентификуваат гените специфични за патогенот како основа за пронао?а?е на антигени кои резултираат со
имунолошки
одговор против патогените соеви, но не и комензалите.
[41]
Во ма? 2019 година, користе??и го Општиот геномски сет, група во
Обединетото Кралство
и
Австрали?а
, секвенционирале ил?адници општо собрани изолати на
стрептокока од групата А
, обезбедува??и потенци?ални цели за разво? на вакцина против патогенот, исто така познат како
S. pyogenes
.
[42]
 Човечки
локуси
на рецептор на Т-клетка (H, горе) и глувците (M, доле) се споредувани, со елементи на рецептор на Т-клетка во црвено, гени без рецептор на Т-клетка во виолетова и V сегменти во портокалова, други елементи на рецептор на Т-клетка во црвено. M6A, наводна метилтрансфераза; ZNF, белковината цинков прст; OR, миризливи рецепторни гени; DAD1, бранител од клеточна смрт; Местата на специфичните видови, обработени
псевдогени
се прикажани со сиви триаголници. Поврзано пристапни броеви на GenBank AE000658-62. Изменето според Глузман и колегите, 2001 година.
[43]
Човечки
локуси
на рецептор на Т-клетка (H, горе) и глувците (M, доле) се споредувани, со елементи на рецептор на Т-клетка во црвено, гени без рецептор на Т-клетка во виолетова и V сегменти во портокалова, други елементи на рецептор на Т-клетка во црвено. M6A, наводна метилтрансфераза; ZNF, белковината цинков прст; OR, миризливи рецепторни гени; DAD1, бранител од клеточна смрт; Местата на специфичните видови, обработени
псевдогени
се прикажани со сиви триаголници. Поврзано пристапни броеви на GenBank AE000658-62. Изменето според Глузман и колегите, 2001 година.
[43]
Моделни глувци во имунологи?ата
[
уреди
|
уреди извор
]
Т-клетките
(исто така познати како Т-лимфоцити или тимоцити) се
имунолошки клетки
кои растат од
матични клетки
во
коскената срцевина
. Тие помагаат да биде заштитено телото од инфекции и може да помогне во борбата против
ракот
. Поради нивната морфолошка,
физиолошка
и
генетска
сличност со
лу?ето
, глувците и
стаорците
долго време се претпочитани видови за биомедицински истражувачки животински модели. Споредбеното медицинско истражува?е е изградено на способноста да бидат користени информации од еден
вид
за да бидат разбрани истите постапки во друг вид. Може да бидат добиени нови сознани?а за молекуларните патишта со споредува?е на Т-клетките на човекот и глушецот и нивните ефекти врз
имунолошкиот систем
користе??и споредбена геномика. Со цел да ги разбере неговите рецептори на Т-клетки и нивните гени, Глусман спроведе истражува?е за секвенционира?е на локусите на рецепторот на Т-клетките на човекот и глувчето. Гените со рецептори на Т-клетки се добро познати и служат како значаен ресурс за поддршка на
функционалната геномика
и разбира?е како гените и ме?угенските региони на геномот придонесуваат за биолошките постапки.
[43]
Имуните рецептори на Т-клетките се важни за да биде види светот на патогени во клеточниот имунолошки систем. Една од причините за секвенционира?е на
локусите
со рецептори на Т-клетки на човекот и глушецот било да бидат совпаднати низите на ортологното семе?ство на гени и да бидат откриени зачуваните области користе??и споредбена геномика. Било сметано дека тие ?е одразуваат два вида биолошки информации: (1) егзони и (2) регулаторни низи. Всушност, поголемиот дел од егзоните V, D, J и C може да бидат идентификувани во ово? метод. Променливите региони се кодирани од пове?е уникатни ДНК елементи кои се преуредени и поврзани за време на диференци?аци?ата на Т-клетките (рецептор на Т-клетка): променлива (V), разновидност (D) и спо?увачки (J) елементи за и
полипептидите
; и елементите V и J за полипептидите. [Слика 1] Сепак, биле прикажани неколку кратки некодирачки зачувани блокови на
геномот
. И човечките и мотивите на глушецот се во голема мера групирани во 200 bp [Слика 2], познатите 3' засилувачи во рецепторот на Т-клетка биле идентификувани и зачувана област од 100 bp во интронот J на глувчето последователно била покажана дека има регулаторна функци?а.
 [Слика 2] Генска структура на генските сегменти V, D, J и C на човечот (горе) и глушецот (долу). Стрелките ?а претставуваат насоката на транскрипци?а на секо? ген со рецептор на Т-клетка. Квадратите и круговите претставуваат движе?е во директна и обратна насока. Изменето според Глузман и колегите, 2001 година.
[43]
[Слика 2] Генска структура на генските сегменти V, D, J и C на човечот (горе) и глушецот (долу). Стрелките ?а претставуваат насоката на транскрипци?а на секо? ген со рецептор на Т-клетка. Квадратите и круговите претставуваат движе?е во директна и обратна насока. Изменето според Глузман и колегите, 2001 година.
[43]
Споредбите на геномските низи во секо?а физичко место или нао?а?е на специфичен ген на хромозомот и ме?у видовите овозможуваат истражува?е на други механизми и други регулаторни сигнали. Некои предлагаат нови хипотези за еволуци?ата на рецептори на Т-клетки, кои треба да бидат тестирани (и подобрени) во споредба со генскиот комплемент на рецепторот на Т-клетка на другите видови
'рбетници
. Споредбеното геномско истражува?е на лу?ето и глувците очигледно ?е овозможи открива?е и прибележува?е на многу други гени, како и идентификува?е ка? други видови за регулаторни низи.
[43]
Споредбената геномика, исто така, отвора нови патишта во други области на истражува?е. Како што технологи?ата за
секвенционира?е на ДНК
стана подостапна, бро?от на секвенционирани геноми расте. Со зголемениот резервоар на достапни геномски податоци, порасна и мо?та на споредбените геномски заклучоци.
Забележителен случа? на оваа зголемена мо? е прона?ден во неодамнешното истражува?е ка?
приматите
. Споредбените геномски методи им овозможило на истражувачите да собираат информации за
генетската вари?аци?а
, диференци?алното
генско изразува?е
и
еволутивната
динамика ка? приматите кои биле незабележливи со користе?е на претходни податоци и методи.
[44]
Проект за геномот на големиот човеколик ма?мун
[
уреди
|
уреди извор
]
Проектот за геномот на големиот човеколик ма?мун користел споредбени геномски методи за да ги истражи
генетските вари?ации
во однос на шесте
видови
големи човеколики ма?муни
, пронао?а??и здрави нивоа на вари?аци?а во нивниот
генски фонд
и покра? намалува?ето на големината на населението.
[45]
Друга студи?а покажала дека моделите на метилаци?а на
ДНК
, кои се познат механизам за регулира?е на
генското изразува?е
, се разликуваат во префронталниот кортекс на
лу?ето
наспроти
шимпанзата
и ?а вмешани оваа разлика во еволутивната дивергенци?а на двата вида.
[46]
- ↑
?Dynamics of genome rearrangement in bacterial populations“
.
PLOS Genetics
.
4
(7): e1000128. ?ули 2008.
doi
:
10.1371/journal.pgen.1000128
.
PMC
2483231
.
PMID
18650965
.

- ↑
2,0
2,1
2,2
?Comparative Genomics“
.
Nature Education Knowledge
.
3
(10): 13. 2010.
- ↑
3,0
3,1
Xia X (2013).
Comparative Genomics
. SpringerBriefs in Genetics. Heidelberg: Springer.
doi
:
10.1007/978-3-642-37146-2
.
ISBN
978-3-642-37145-5
.
- ↑
4,0
4,1
Russel PJ, Hertz PE, McMillan B (2011).
Biology: The Dynamic Science
(2. изд.). Belmont, CA: Brooks/Cole. стр. 409?410.
- ↑
Primrose SB, Twyman RM (2003).
Principles of Genome Analysis and Genomics
(3. изд.). Malden, MA: Blackwell Publishing.
ISBN
9781405101202
.
- ↑
?Comparative genomics“
.
PLOS Biology
.
1
(2): E58. ноември 2003.
doi
:
10.1371/journal.pbio.0000058
.
PMC
261895
.
PMID
14624258
.
- ↑
?Comparative genomics and the study of evolution by natural selection“.
Molecular Ecology
.
17
(21): 4586?4596. ноември 2008.
doi
:
10.1111/j.1365-294X.2008.03954.x
.
PMID
19140982
.
- ↑
8,0
8,1
8,2
8,3
Koonin EV, Galperin MY (2003).
Sequence - Evolution - Function: Computational approaches in comparative genomics
. Dordrecht: Springer Science+Business Media.
- ↑
9,0
9,1
?Pathogen comparative genomics in the next-generation sequencing era: genome alignments, pangenomics and metagenomics“.
Briefings in Functional Genomics
.
10
(6): 322?333. ноември 2011.
doi
:
10.1093/bfgp/elr042
.
PMID
22199376
.
- ↑
?Similarity in gene organization and homology between proteins of animal picornaviruses and a plant comovirus suggest common ancestry of these virus families“
.
Nucleic Acids Research
.
12
(18): 7251?7267. септември 1984.
doi
:
10.1093/nar/12.18.7251
.
PMC
320155
.
PMID
6384934
.
- ↑
?DNA sequence of the herpes simplex virus type 1 gene encoding glycoprotein gH, and identification of homologues in the genomes of varicella-zoster virus and Epstein-Barr virus“
.
Nucleic Acids Research
.
14
(10): 4281?4292. ма? 1986.
doi
:
10.1093/nar/14.10.4281
.
PMC
339861
.
PMID
3012465
.
- ↑
?Whole-genome random sequencing and assembly of Haemophilus influenzae Rd“.
Science
.
269
(5223): 496?512. ?ули 1995.
Bibcode
:
1995Sci...269..496F
.
doi
:
10.1126/science.7542800
.
PMID
7542800
.
CS1-одржува?е: display-автори (
link
)
- ↑
?The minimal gene complement of Mycoplasma genitalium“.
Science
.
270
(5235): 397?403. октомври 1995.
Bibcode
:
1995Sci...270..397F
.
doi
:
10.1126/science.270.5235.397
.
PMID
7569993
.
CS1-одржува?е: display-автори (
link
)
- ↑
?Life with 6000 genes“.
Science
.
274
(5287): 546, 563?546, 567. октомври 1996.
Bibcode
:
1996Sci...274..546G
.
doi
:
10.1126/science.274.5287.546
.
PMID
8849441
.
CS1-одржува?е: display-автори (
link
)
- ↑
?Genome sequence of the nematode C. elegans: a platform for investigating biology“.
Science
.
282
(5396): 2012?2018. декември 1998.
Bibcode
:
1998Sci...282.2012.
.
doi
:
10.1126/science.282.5396.2012
.
PMID
9851916
.
- ↑
?The genome sequence of Drosophila melanogaster“.
Science
.
287
(5461): 2185?2195. март 2000.
Bibcode
:
2000Sci...287.2185.
.
CiteSeerX
10.1.1.549.8639
.
doi
:
10.1126/science.287.5461.2185
.
PMID
10731132
.
CS1-одржува?е: display-автори (
link
)
- ↑
?Comparative genomics of the eukaryotes“
.
Science
.
287
(5461): 2204?2215. март 2000.
Bibcode
:
2000Sci...287.2204.
.
doi
:
10.1126/science.287.5461.2204
.
PMC
2754258
.
PMID
10731134
.
CS1-одржува?е: display-автори (
link
)
- ↑
?Human and mouse gene structure: comparative analysis and application to exon prediction“
.
Genome Research
.
10
(7): 950?958. ?ули 2000.
doi
:
10.1101/gr.10.7.950
.
PMC
310911
.
PMID
10899144
.
- ↑
?Comparative genomics: genome-wide analysis in metazoan eukaryotes“.
Nature Reviews. Genetics
.
4
(4): 251?262. април 2003.
doi
:
10.1038/nrg1043
.
PMID
12671656
.
- ↑
?The genome sequence of Caenorhabditis briggsae: a platform for comparative genomics“
.
PLOS Biology
.
1
(2): E45. ноември 2003.
doi
:
10.1371/journal.pbio.0000045
.
PMC
261899
.
PMID
14624247
.
CS1-одржува?е: display-автори (
link
)
- ↑
?Newly Sequenced Worm a Boon for Worm Biologists“
.
PLOS Biology
.
1
(2): e4. 2003.
doi
:
10.1371/journal.pbio.0000044
.
PMC
261884
.
- ↑
Cristianini N, Hahn M (2006).
Introduction to Computational Genomics
. Cambridge University Press.
ISBN
978-0-521-67191-0
.
- ↑
?An alignment-free method to find and visualise rearrangements between pairs of DNA sequences“
.
Scientific Reports
.
5
: 10203. ма? 2015.
Bibcode
:
2015NatSR...510203P
.
doi
:
10.1038/srep10203
.
PMC
4434998
.
PMID
25984837
.
- ↑
?Ten simple rules for developing a short bioinformatics training course“
.
PLOS Computational Biology
.
7
(10): e1002245. октомври 2011.
Bibcode
:
2011PLSCB...7E2245V
.
doi
:
10.1371/journal.pcbi.1002245
.
PMC
3203054
.
PMID
22046119
.
CS1-одржува?е: display-автори (
link
)
- ↑
25,0
25,1
?Evolution of the ancestral mammalian karyotype and syntenic regions“
.
Proceedings of the National Academy of Sciences of the United States of America
.
119
(40): e2209139119. октомври 2022.
doi
:
10.1073/pnas.2209139119
.
PMC
9550189
.
PMID
36161960
.
CS1-одржува?е: display-автори (
link
)
- ↑
Altschul SF, Pop M (2017).
?Sequence Alignment“
. Во Rosen KH, Shier DR, Goddard W (уред.).
Handbook of Discrete and Combinatorial Mathematics
(2nd. изд.). Boca Raton (FL): CRC Press/Taylor & Francis.
ISBN
978-1-58488-780-5
.
PMID
29206392
. Посетено на
2022-12-18
.
- ↑
Prjibelski AD, Korobeynikov AI, Lapidus AL (2019-01-01). ?Sequence Analysis“. Во Ranganathan S, Gribskov M, Nakai K, Schonbach C (уред.).
Encyclopedia of Bioinformatics and Computational Biology
(англиски). Oxford: Academic Press. стр. 292?322.
doi
:
10.1016/b978-0-12-809633-8.20106-4
.
ISBN
978-0-12-811432-2
.
- ↑
?Comparative genomics: methods and applications“.
Die Naturwissenschaften
.
91
(9): 405?421. септември 2004.
doi
:
10.1007/s00114-004-0542-8
.
PMID
15278216
.
- ↑
?Inferring synteny between genome assemblies: a systematic evaluation“
.
BMC Bioinformatics
.
19
(1): 26. ?ануари 2018.
doi
:
10.1186/s12859-018-2026-4
.
PMC
5791376
.
PMID
29382321
.
- ↑
Duran C, Edwards D, Batley J (2009). ?Genetic Maps and the Use of Synteny“.
Plant Genomics
. Methods in Molecular Biology.
513
. стр. 41?55.
doi
:
10.1007/978-1-59745-427-8_3
.
ISBN
978-1-58829-997-0
.
PMID
19347649
.
- ↑
31,0
31,1
31,2
Bergman NH (2007). Bergman NH (уред.).
Comparative Genomics: Volumes 1 and 2
. Totowa, New Jersey: Humana Press.
ISBN
978-193411-537-4
.
PMID
21250292
.
- ↑
?UCSC Browser“
.
- ↑
?Ensembl Genome Browser“
. Архивирано од
изворникот
на 2013-10-21.
- ↑
?Map Viewer“
.
- ↑
?VISTA tools“
.
- ↑
?The Bluejay genome browser“.
Current Protocols in Bioinformatics
. John Wiley & Sons, Inc.
37
: Unit10.9. март 2012.
doi
:
10.1002/0471250953.bi1009s37
.
ISBN
9780471250951
.
PMID
22389011
.
- ↑
?Genome-wide association studies of 14 agronomic traits in rice landraces“.
Nature Genetics
.
42
(11): 961?967. ноември 2010.
doi
:
10.1038/ng.695
.
PMID
20972439
.
CS1-одржува?е: display-автори (
link
)
- ↑
?Crop genomics: advances and applications“.
Nature Reviews. Genetics
.
13
(2): 85?96. декември 2011.
doi
:
10.1038/nrg3097
.
PMID
22207165
.
- ↑
?Developing vaccines in the era of genomics: a decade of reverse vaccinology“.
Clinical Microbiology and Infection
. 18 Suppl 5 (SI): 109?116. октомври 2012.
doi
:
10.1111/j.1469-0691.2012.03939.x
.
PMID
22882709
.
- ↑
?Identification of a universal Group B streptococcus vaccine by multiple genome screen“
.
Science
.
309
(5731): 148?150. ?ули 2005.
Bibcode
:
2005Sci...309..148M
.
doi
:
10.1126/science.1109869
.
PMC
1351092
.
PMID
15994562
.
CS1-одржува?е: display-автори (
link
)
- ↑
?The pangenome structure of Escherichia coli: comparative genomic analysis of E. coli commensal and pathogenic isolates“
.
Journal of Bacteriology
.
190
(20): 6881?6893. октомври 2008.
doi
:
10.1128/JB.00619-08
.
PMC
2566221
.
PMID
18676672
.
CS1-одржува?е: display-автори (
link
)
- ↑
?Group a Streptococcus Vaccine Target Candidates Identified from Global Genome Set“
. 28 ма? 2019.
- ↑
43,0
43,1
43,2
43,3
?Comparative genomics of the human and mouse T cell receptor loci“.
Immunity
.
15
(3): 337?349. септември 2001.
doi
:
10.1016/s1074-7613(01)00200-x
.
PMID
11567625
.
CS1-одржува?е: display-автори (
link
)
- ↑
?Comparative primate genomics: emerging patterns of genome content and dynamics“
.
Nature Reviews. Genetics
.
15
(5): 347?359. ма? 2014.
doi
:
10.1038/nrg3707
.
PMC
4113315
.
PMID
24709753
.
- ↑
?Great ape genetic diversity and population history“
.
Nature
.
499
(7459): 471?475. ?ули 2013.
Bibcode
:
2013Natur.499..471P
.
doi
:
10.1038/nature12228
.
PMC
3822165
.
PMID
23823723
.
CS1-одржува?е: display-автори (
link
)
- ↑
?Divergent whole-genome methylation maps of human and chimpanzee brains reveal epigenetic basis of human regulatory evolution“
.
American Journal of Human Genetics
.
91
(3): 455?465. септември 2012.
doi
:
10.1016/j.ajhg.2012.07.024
.
PMC
3511995
.
PMID
22922032
.
- Kellis M, Patterson N, Endrizzi M, Birren B, Lander ES (ма? 2003). ?Sequencing and comparison of yeast species to identify genes and regulatory elements“.
Nature
.
423
(6937): 241?254.
Bibcode
:
2003Natur.423..241K
.
doi
:
10.1038/nature01644
.
PMID
12748633
.
S2CID
1530261
.
- Cliften P, Sudarsanam P, Desikan A, Fulton L, Fulton B, Majors J, и др. (?ули 2003). ?Finding functional features in Saccharomyces genomes by phylogenetic footprinting“.
Science
.
301
(5629): 71?76.
Bibcode
:
2003Sci...301...71C
.
doi
:
10.1126/science.1084337
.
PMID
12775844
.
S2CID
1305166
.
- Boffelli D, McAuliffe J, Ovcharenko D, Lewis KD, Ovcharenko I, Pachter L, Rubin EM (февруари 2003).
?Phylogenetic shadowing of primate sequences to find functional regions of the human genome“
.
Science
.
299
(5611): 1391?1394.
doi
:
10.1126/science.1081331
.
PMID
12610304
.
S2CID
17217612
.
- Dujon B, Sherman D, Fischer G, Durrens P, Casaregola S, Lafontaine I, и др. (?ули 2004). ?Genome evolution in yeasts“.
Nature
.
430
(6995): 35?44.
Bibcode
:
2004Natur.430...35D
.
doi
:
10.1038/nature02579
.
PMID
15229592
.
S2CID
4399964
.
- Filipski A, Kumar S (2005). ?Comparative genomics in eukaryotes“. Во Gregory TR (уред.).
The Evolution of the Genome
. San Diego: Elsevier. стр. 521?583.
- Gregory TR, DeSalle R (2005). ?Comparative genomics in prokaryotes“. Во Gregory TR (уред.).
The Evolution of the Genome
. San Diego: Elsevier. стр. 585?675.
- Xie X, Lu J, Kulbokas EJ, Golub TR, Mootha V, Lindblad-Toh K, и др. (март 2005).
?Systematic discovery of regulatory motifs in human promoters and 3' UTRs by comparison of several mammals“
.
Nature
.
434
(7031): 338?345.
Bibcode
:
2005Natur.434..338X
.
doi
:
10.1038/nature03441
.
PMC
2923337
.
PMID
15735639
.
- Champ PC, Binnewies TT, Nielsen N, Zinman G, Kiil K, Wu H, и др. (март 2006). ?Genome update: purine strand bias in 280 bacterial genomes“.
Microbiology
.
152
(Pt 3): 579?583.
doi
:
10.1099/mic.0.28637-0
.
PMID
16514138
.
- Kumar L, Breakspear A, Kistler C, Ma LJ, Xie X (март 2010).
?Systematic discovery of regulatory motifs in Fusarium graminearum by comparing four Fusarium genomes“
.
BMC Genomics
.
11
: 208.
doi
:
10.1186/1471-2164-11-208
.
PMC
2853525
.
PMID
20346147
.
- Batzoglou S, Pachter L, Mesirov JP, Berger B, Lander ES (?ули 2000).
?Human and mouse gene structure: comparative analysis and application to exon prediction“
.
Genome Research
.
10
(7): 950?958.
doi
:
10.1101/gr.10.7.950
.
PMC
310911
.
PMID
10899144
.