| Klassifikation nach
ICD-10
|
| D57.0
|
Sichelzellenanamie mit Krisen
Hb-SS-Krankheit mit Krisen
|
| D57.1
|
Sichelzellenanamie ohne Krisen
|
| D57.2
|
Doppelt heterozygote Sichelzellenkrankheit
|
| D57.3
|
Sichelzellen-Erbanlage
Heterozygotes Hamoglobin S
|
| D57.8
|
Sonstige Sichelzellenkrankheiten
|
| ICD-10 online (WHO-Version 2019)
|
Die
Sichelzellanamie
(auch
Sichelzellkrankheit
oder
Drepanozytose
) ist eine
erbliche Erkrankung
der roten Blutkorperchen (
Erythrozyten
). Sie gehort zur Gruppe der
Hamoglobinopathien
(Storungen des
Hamoglobins
) und fuhrt zu einer
korpuskularen
hamolytischen Anamie
. Bei den Betroffenen liegt eine Mutation der β-Kette des Hamoglobins vor. Es konnen entweder alle β-Ketten betroffen sein (schwere,
homozygote
Form) oder nur ein Teil (mildere,
heterozygote
Form).
Die Krankheit tritt vor allem bei Personen aus
Subsahara-Afrika
und deren Nachfahren,
[1]
aber auch in Teilen des Mittelmeerraums und des Nahen Ostens bis Indien auf und wurde durch Migration global verbreitet.
[2]
Sie ist nach wie vor in den Entwicklungslandern mit einer hohen
Mortalitat
verbunden.
[2]
Die Krankheit wurde 1910 von
James Herrick
[3]
und
Ernest E. Irons
[3]
bei einem Patienten aus der Karibik beschrieben und die Bezeichnung Sichelzellenanamie wurde zuerst von
Verne R. Mason
1922 benutzt.
[4]
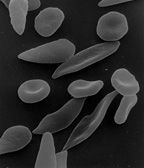 Sichelformige rote Blutkorperchen
Sichelformige rote Blutkorperchen
 Blutausstrich
bei Sichelzellanamie
Blutausstrich
bei Sichelzellanamie
Die Betroffenen bilden ein abnormes Hamoglobin (
Sichelzell-Hamoglobin
,
HbS
), das bei
Sauerstoffmangel
zur Bildung von
Fibrillen
neigt. Dabei verformen sich die roten Blutzellen durch die enthaltenen Fasern zu sichelformigen Gebilden, verklumpen miteinander und verstopfen kleine Blutgefaße,
[5]
wodurch eine
Entzundung
entsteht.
[6]
Durch die Verklumpung und Gefaßverstopfung kann es bei der homozygoten Form zu anfallsartigen schmerzhaften, zum Teil lebensbedrohlichen Durchblutungsstorungen (
Sichelzellkrisen
) kommen,
[7]
die unter anderem zu venosen
Thrombosen
fuhren konnen.
[8]
Heterozygot Betroffene, bei denen nur eines der beiden Hamoglobin-Gene verandert ist, sind vor den schweren Verlaufsformen der
Malaria
geschutzt. Dadurch ist das mutierte Hamoglobin-Gen in Malariagebieten relativ verbreitet.
Die Zerstorung roter Blutkorperchen fuhrt zu einer schweren chronischen Blutarmut (
hamolytische Anamie
). Aufgrund der Neigung des
Hamoglobin S
zur
Polymerisation
und der sichelformigen Deformierung der Erythrozyten kommt es zu Verschlussen kleiner
Arterien
mit
rezidivierenden
Durchblutungsstorungen
. Dies fuhrt zu starken Schmerzen und Schaden in multiplen Organsystemen:
Gehirn
(
ischamischer Schlaganfall
),
Milzinfarkt
,
Lunge
(
Lungenentzundung
,
pulmonale Hypertonie
),
Auge
,
Herz
- und
Nierenversagen
,
Muskel
,
Knochen
(
Osteonekrose
) oder
Priapismus
. Die
Lebenserwartung
ist vermindert.
[9]
Eine
Glomerulopathie
mit
Hyperfiltration
tritt bei bis zu einem Drittel der Patienten mit homozygotem Phanotyp schon in der Kindheit auf. Schaden im Bereich des
Nierenmarks
fuhren zu
Papillennekrosen
, Verlust der Konzentrationsfahigkeit der Nieren und
blutigem Urin (Makrohamaturie)
. Schaden im Bereich der
Nierenkorperchen
(Glomeruli) fuhren zu vermehrter Eiweißausscheidung im Urin (
Mikro-
und
Makroalbuminurie
,
nephrotisches Syndrom
). Bei der
feingeweblichen Untersuchung
ist die vorherrschende glomerulare Schadigung die
fokal segmentale Glomerulosklerose
. Bei bis zu einem Drittel der Patienten kommt es in den ersten Lebensjahrzehnten zum Auftreten einer
Proteinurie
, in funf Prozent zu einem terminalen Nierenversagen.
[10]
[11]
Nur homozygote Trager des Sichelzellgens zeigen diese starke Auspragung der Krankheit, bei der das gesamte Hamoglobin abnormes Sichelzellhamoglobin (irregulares Hamoglobin) ist. In heterozygoten Tragern ist nur etwa ein Prozent aller Erythrozyten deformiert. Die Symptome verschlimmern sich erheblich, wenn die Patienten korperlich stark aktiv sind oder sich in großen Hohen befinden. Dies liegt daran, dass sich die Sichelform der Erythrozyten bei niedrigem
Sauerstoffpartialdruck
bildet, weil unter diesen Bedingungen das Hamoglobin faserig ausfallt (die Loslichkeit von Hamoglobin bei Sichelzellanamie ist 25-fach geringer als die Loslichkeit von normalem Hamoglobin).
Etwa ab dem sechsten Lebensmonat, wenn der Abbau des fetalen
Hamoglobins
bereits weit fortgeschritten ist, konnen erstmals Symptome auftreten. Sie außern sich dann meist in einer sogenannten Sichelzellkrise: Durch außere Einflusse wie Anstrengung sinkt der Sauerstoffpartialdruck im Blut, die
Sichelzellen
werden
hamolytisch
.
Aufgrund einer Punktmutation im HBB-Gen (c.20A>T) auf
Chromosom
11 ist bei der Sichelzellenanamie an der Position sechs der
β-Globin
-
Protein-Untereinheit
des Hamoglobins die Aminosaure
Glutaminsaure
durch
Valin
ersetzt.
[12]
Die Bezeichnung dieser Variante in
offizieller genetischer Nomenklatur
lautet HBB-p.E6V. Die betroffenen Erythrozyten verformen sich bei abnehmendem Sauerstoffpartialdruck sichelformig, verfangen sich leicht in den
Kapillaren
und
lysieren
uberdies sehr schnell. Durch die
Hamolyse
werden
Hamoglobin
,
Arginase
und freie
Sauerstoffradikale
freigesetzt. Freies Hamoglobin bindet
Stickstoffmonoxid
etwa 1000-mal starker als intrazellulares, und Arginase verwandelt Stickstoffmonoxid zu Nitrit und Nitrat. Stickstoffmonoxid ist der wichtigste
Vasodilatator
, und die Konzentrationsabnahme fuhrt zur Gefaßverengung und somit zu Durchblutungsstorungen.
Das Sichelzellenhamoglobin wird als HbS bezeichnet im Gegensatz zum HbA, dem normalen Hamoglobin des erwachsenen Menschen. Heterozygote Trager des Merkmals bilden neben dem HbS auch HbA in ausreichender Menge, um die Funktion der Erythrozyten bei diesen Menschen weitgehend aufrechtzuerhalten.
Am Beispiel der Sichelzellenanamie wurde erstmals der Zusammenhang eines Defekts eines Molekuls mit einer Krankheit nachgewiesen, in einer beruhmten Arbeit von
Linus Pauling
,
Harvey Itano
und
Seymour Jonathan Singer
von 1949.
[13]
[14]
Der Unterschied des Hamoglobins beider roter Blutkorperchen zeigte sich in der Gel-
Elektrophorese
, die Itano durchfuhrte. Die Autoren vermuteten schon, dass Unterschiede in den Aminosauren vorlagen, was
Vernon Ingram
1956 bestatigte, der auch zeigte, dass der Unterschied im Austausch genau einer Aminosaure bestand. Die Vererbungsmuster der Krankheit klarte ebenfalls 1949
James V. Neel
(1915?2000).
Sichelzellenanamie ist eine
autosomal
kodominante
Erbkrankheit
.
- Das Erbgut eines Gesunden enthalt die beiden unvollstandig dominanten (auf molekularem Niveau sind A und S kodominant)
Allele
(AA) fur das Hamoglobin A. Seine roten Blutkorperchen sind stets elastisch.
- Ein
Ubertrager (Konduktor)
mit dem
Genotyp
AS (=
heterozygot
) enthalt sowohl das Allel A als auch das mutierte Allel S, welches das veranderte Hamoglobin S verursacht. Seine roten Blutkorperchen enthalten HbA und HbS im Verhaltnis 1:1. Unter Normalbedingungen zeigen die roten Blutkorperchen keine Veranderungen, die Krankheit kommt nicht zum Ausbruch. Erst unter sehr starkem Sauerstoffmangel verformen sich die roten Blutkorperchen zu sichelformigen Gebilden, was die Durchblutung der Organe beeintrachtigt.
- Ein Trager des Genotyps SS (
homozygot
) stellt nur das veranderte HbS her. Schon unter physiologischem Sauerstoffmangel, wie er z. B. in den Kapillaren sauerstoffverbrauchender Organe herrscht, kommt es zu einer starken Verformung der roten Blutkorperchen. Sie verlieren ihre Elastizitat und verhaken leicht miteinander. Dadurch kommt es zu einem Verschluss der Kapillaren. Unter normalen Bedingungen ist das Hamoglobin in den roten Blutkorperchen stets fein verteilt. Bei abnehmendem
pH-Wert
und Sauerstoffgehalt des Blutes kommt es beim HbS zu einer Verklumpung der Hamoglobinmolekule zu stabchenformigen, kristallinen Gebilden. Dadurch wird der Erythrozyt sichelformig verformt und verliert seine Elastizitat.
Die Diagnose der Sichelzellanamie erfolgt zunachst
anamnestisch
, wobei die Herkunft und andere Falle der Erkrankung in der Familie erfragt werden sollten; danach klinisch anhand der gebotenen Symptome und schließlich im Labor, wo in einem Blutbild eine hamolytische Anamie erscheinen kann und bei Untersuchung des Blutes unter dem Mikroskop die typisch geformten Drepanozyten erscheinen - insbesondere, wenn das Blut fur 24 h unter Sauerstoffabschluss gelagert wurde (Sichelzelltest). Außerdem kann eine
Elektrophorese des Hamoglobins
die veranderten Molekule beweisend identifizieren. Schließlich kann auch der Genabschnitt fur das Hamoglobin in einer Restriktionsanalyse untersucht werden und die Punktmutation auf der Ebene der DNA aufzeigen.
Sind die Genotypen der Eltern bekannt, kann die
Wahrscheinlichkeit
berechnet werden, mit der die Sichelzellenanamie bei einem Kind auftritt:
Genotyp
der Eltern
|
Genotypen
der Kinder
|
Vererbungswahrscheinlichkeiten und Phanotyp der Kinder
|
| AA x AA
|
AA
|
100 %
|
gesunde Kinder
|
| AA × AS
|
AA
|
0
50 %
|
Wahrscheinlichkeit, dass ein Kind gesund ist
|
| AS
|
0
50 %
|
Wahrscheinlichkeit, dass ein Kind heterozygoter HbS-Trager wird
|
| AS × AS
|
AA
|
0
25 %
|
Wahrscheinlichkeit, dass ein Kind gesund ist
|
| AS
|
0
50 %
|
Wahrscheinlichkeit, dass ein Kind heterozygoter HbS-Trager wird
|
| SS
|
0
25 %
|
Wahrscheinlichkeit, dass ein Kind von der schwereren, homozygoten Form betroffen ist
|
| AA × SS
|
AS
|
100 %
|
alle Kinder werden heterozygote HbS-Trager
|
| AS × SS
|
AS
|
0
50 %
|
Wahrscheinlichkeit, dass ein Kind heterozygoter HbS-Trager wird
|
| SS
|
0
50 %
|
Wahrscheinlichkeit, dass ein Kind von der schwereren, homozygoten Form betroffen ist
|
| SS × SS
|
SS
|
100 %
|
Die Kinder haben mit Sicherheit die schwerere, homozygote Form
|
Das zu untersuchende Blut wird mit
EDTA
versetzt und uber 24 Stunden unter Sauerstoffabschluss gelagert. Durch den eintretenden Sauerstoffmangel in den Erythrozyten entstehen die Sichelformen der Zellen, die im Mikroskop bei 40-facher Vergroßerung gut erkannt werden konnen. Zusatzlich kann durch Zugabe von
Natriummetabisulfit
[15]
der Effekt der Sichelzellbildung beschleunigt werden.
[16]
 Hamoglobin-Gelelektrophorese: Hetero- und homozygotes Vorliegen einer Variante (hier a) lassen sich unterscheiden.
Hamoglobin-Gelelektrophorese: Hetero- und homozygotes Vorliegen einer Variante (hier a) lassen sich unterscheiden.
Da erst unter extremem Sauerstoffmangel eine Veranderung der roten Blutkorperchen von Ubertragern (Genotyp AS) auftritt, lasst sich durch Untersuchung der roten Blutkorperchen unter dem
Mikroskop
der Genotyp AA nicht vom Genotyp AS unterscheiden. Dagegen lasst sich mit Hilfe der
Hamoglobin-Elektrophorese
eindeutig der Genotyp bestimmen: Dazu wird den Probanden Blut entnommen und aufbereitet, bis reines Hamoglobin vorliegt. Im
elektrischen Feld
wandern die beiden Hamoglobinsorten unterschiedlich weit, da HbS aufgrund seiner geanderten Proteinstruktur ein anderes Wanderungsverhalten zeigt. Auch klinisch relevante Kombinationsformen, wie z. B. aus HbS und
Hamoglobin C
, die s.g. HbSC-Krankheit oder die Kombination aus HbS und Hamoglobin E (HbE) konnen mit der Hb-Elektrophorese unterschieden werden.
[17]
Die ursachliche Mutation im HBB-Gen (c.20A>T) kann mittels molekularbiologischer Untersuchungsverfahren nachgewiesen werden.
[12]
Die
Sequenzierung
des HBB-Gens verdrangt hierbei zunehmend andere Verfahren wie bspw. die
Restriktionsanalyse
.
[18]
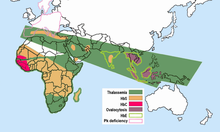 Geographische Verteilung der haufigsten Hamoglobinopathien. Hamoglobin S ist in Gelb dargestellt.
Geographische Verteilung der haufigsten Hamoglobinopathien. Hamoglobin S ist in Gelb dargestellt.
Auffallend ist, dass in Gebieten der
Malaria
das Sichelzellenallel relativ haufig ist. Daraus wurde geschlussfolgert, dass es gegen Malaria eine
Resistenz
verleiht, sodass die gesunden Ubertrager (AS) des Sichelzellenallels in diesen Gebieten einen
Selektionsvorteil
(den sogenannten
Heterozygotenvorteil
) gegenuber denen ohne Sichelzellenallel (Genotyp AA), die eher an Malaria sterben, und auch gegenuber den Sichelzellerkrankten (Genotyp SS) haben, die vorzeitig an Sichelzellenanamie sterben. In Afrika gibt es beispielsweise Gegenden, in denen fast ein Drittel der Bevolkerung heterozygot fur dieses Merkmal ist. In den anderen Weltgegenden kommt das Sichelzellenallel praktisch nicht vor, da hier dieser Selektionsvorteil aufgrund der fehlenden Malaria nicht wirksam ist. In Deutschland werden jahrlich zwischen 500 und 1100 Falle von Malaria festgestellt.
[19]
Etwa 85 % der homozygoten Trager stammen aus Afrika.
[20]
Verlassliche Daten zur Haufigkeit in Deutschland liegen nicht vor. Die Gesamtzahl der in Deutschland lebenden Patienten mit Sichelzellkrankheit wurde fur das Jahr 2017 auf mindestens 2000 geschatzt.
[21]
 H
+
-Ionen begunstigen die Sauerstoffabgabe und die defekten β-Ketten konnen sich verbinden. Es kommt zur Sichelform.
H
+
-Ionen begunstigen die Sauerstoffabgabe und die defekten β-Ketten konnen sich verbinden. Es kommt zur Sichelform.
Moglicherweise besteht ein Selektionsvorteil von heterozygoten Tragern bei Infektionen mit
Malaria
.
[22]
Der Malaria-Erreger wird wahrend eines Teiles seines Entwicklungszyklus an oder in den Erythrozyten transportiert. Das Hamoglobin von Menschen mit der heterozygoten Form des HbS fuhrt durch Verminderung der Sauerstoffsattigung des Hamoglobins unter Extrembedingungen zur sichelartigen Verformung der roten Blutzellen, die dann in der Milz abgebaut werden oder verklumpen und danach zugrunde gehen. Eine Hypothese besagt, diejenigen Zellen, die von
Plasmodien
befallen sind, wurden sich auch ohne diese Druckverminderung schon allein durch den Einfluss der
Merozoiten beziehungsweise der Trophozoiten
verformen, von der Milz als krank erkannt und abgebaut werden.
Eine weitere Hypothese ist die direkte Totung der Parasiten, denn die Sichelzellen bilden vermehrt Sauerstoffradikale. Es entstehen dabei Superoxidanionen und
Wasserstoffperoxid
, und beide Verbindungen sind fur die Parasiten giftig.
[23]
Eine andere Theorie besagt Folgendes: Befallen die Plasmodien, die die Malaria verursachen, Erythrozyten, so setzen die
Mikroben
nach einiger Zeit Sauren als Abfallprodukte ihres Stoffwechsels frei. Das Hamoglobin gibt nun, von H
+
-Ionen angeregt, den Sauerstoff ab (Rechtsverschiebung der
Sauerstoffbindungskurve
). Die Sichelform betrifft aber vor allem die Desoxyform der Erythrozyten. Also werden die befallenen Zellen schnell zu Sichelzellen, und diese werden dann in der Milz samt den Mikroben abgebaut. Daraus erklart sich die Resistenz der Trager der Sichelzellenanamie gegenuber Malaria (siehe Abbildung).
Die letzte Theorie besagt, dass dabei unter der Bildung von Hamoglobinpolymeren Hamin entsteht, das wiederum zur direkten Totung der Parasiten fuhrt.
[24]
Momentan werden Ansatze zur Verstarkung der
Genexpression
von
HbF
in Adoleszenten und Erwachsenen untersucht.
[25]
Hydroxyurea
kann die Bildung von HbF induzieren.
[26]
Rote Blutkorperchen mit hohem Anteil von HbF bilden keine Sichelzellen, werden daher seltener abgebaut und verursachen seltener Verschlusse kleiner Gefaße. Eine Behandlung mit Hydroxyurea kann die Haufigkeit von Gefaßverschlussen vermindern,
[27]
chronische Organschaden mildern und das Uberleben verlangern.
[28]
Dieses konnte kurzlich auch bei Kindern im
Subsahara-Afrika
gezeigt werden.
[29]
Weiterhin wird ein
adoptiver Zelltransfer
untersucht.
[30]
Am 12. Oktober 2016 wurde eine Therapiemoglichkeit basierend auf einer Veranderung der betroffenen Gene mit der
CRISPR/Cas-Methode
veroffentlicht. Mit Hilfe der Genschere ersetzten die Forscher die krankmachende Mutation durch die korrekten DNA-Basen. Es wurden zum ersten Mal ausreichend gesunde Blutzellen erzeugt, um Patienten kunftig mit dieser Methode heilen zu konnen, wie die Forscher um
Jacob Corn
von der
University of California
, Berkeley, berichteten. Es sei noch zu fruh, um von einer praktikablen Losung zu sprechen, jedoch sei der erste Schritt gemacht um die Ursachen zu bekampfen, anstatt eine Symptombehandlung durchzufuhren.
[31]
[32]
Ein weiteres Arzneimittel ist der Sauerstoffaffinitats-Modulator
Voxelotor
.
[33]
Am 12. Oktober 2023 hat die U.S.
Food and Drug Administration
(FDA) zwei Behandlungen der Sichelzellkrankheit fur Patienten ab 12 Jahren mit wiederholten Sichelzellkrisen zugelassen
[34]
. Eine der Behandlungen nutzt patienteneigene
Blutstammzellen
, die dem Patienten entnommen, auf Basis der
CRISPR/Cas-Methode
modifiziert und als Einmaldosis in das
Knochenmark
zuruck transplantiert werden. Vor dem letzten Schritt mussen jedoch verbleibende Stammzellen im Knochenmark mittels
Chemotherapie
entfernt werden.
- Fernando Ferreira Costa, Nicola Conran (Hrsg.):
Sickle Cell Anemia. From Basic Science to Clinical Practice.
Springer 2016
- ↑
Kompaktlexikon Biologie, Spektrum Akademischer Verlag
- ↑
a
b
Frederic Piel, Thomas Williams:
Sickle Cell Anemia ? History and Epidemiology.
In: Fernando Ferreira Costa, Nicola Conran (Hrsg.):
Sickle Cell Anemia. From Basic Science to Clinical Practice
. Springer, 2016, S. 24.
- ↑
a
b
J. B. Herrick:
Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia
. In:
Archives of Internal Medicine
.
Band
6
, 1910,
S.
517?521
,
PMC 2588723
(freier Volltext) – (Reprint).
- ↑
V. R. Mason:
Sickle Cell Anemia
. In:
The Journal of the American Medical Association
.
Band
79
, 1922,
S.
1318?1320
,
doi
:
10.1001/jama.254.14.1955
(Reprint).
- ↑
K. Sun, Y. Xia:
New insights into sickle cell disease: a disease of hypoxia.
In:
Current Opinion in Hematology
.
Band 20, Nummer 3, Mai 2013, S. 215?221.
doi:10.1097/MOH.0b013e32835f55f9
.
PMID 23549375
.
- ↑
E. Sparkenbaugh, R. Pawlinski:
Interplay between coagulation and vascular inflammation in sickle cell disease.
In:
British Journal of Haematology
.
Band 162, Nummer 1, Juli 2013, S. 3?14.
doi:10.1111/bjh.12336
.
PMC 3878906
(freier Volltext).
- ↑
B. E. Gee:
Biologic complexity in sickle cell disease: implications for developing targeted therapeutics.
In:
TheScientificWorldJournal.
2013, S. 694146.
doi:10.1155/2013/694146
.
PMC 3621302
(freier Volltext).
- ↑
M. Y. Lim, K. I. Ataga, N. S. Key:
Hemostatic abnormalities in sickle cell disease.
In:
Current Opinion in Hematology.
Band 20, Nummer 5, September 2013, S. 472?477.
doi:10.1097/MOH.0b013e328363442f
.
PMID 23817169
.
- ↑
S. Sheth, M. Licursi, M. Bhatia:
Sickle cell disease: time for a closer look at treatment options?
In:
British Journal of Haematology
.
Band 162, Nummer 4, August 2013, S. 455?464.
doi:10.1111/bjh.12413
- ↑
Harrisons Innere Medizin, 15. Auflage, S. 1754.
- ↑
Raimund Hirschberg:
Glomerular hyperfiltration in sickle cell disease
. In:
Clinical Journal of the American Society of Nephrology
.
Band
5
,
Nr.
5
, Mai 2010,
S.
748?749
,
doi
:
10.2215/CJN.01340210
.
- ↑
a
b
ClinVar
(Studienverzeichnis):
NM_000518.5(HBB):c.20A>T (p.Glu7Val) AND HEMOGLOBIN S
- ↑
L. Pauling, Harvey A. Itano, S. J. Singer, Ibert C. Wells: Sickle Cell Anemia, a Molecular Disease, Science, Band 110, 1949, S. 543?548.
- ↑
D. Lowe, Das Chemiebuch, Librero 2017, S. 354.
- ↑
Prof. R. Heinz: Transfusionsmedizin und Migration
@1
@2
Vorlage:Toter Link/www.roteskreuz.at
(
Seite nicht mehr abrufbar
, festgestellt im Mai 2024.
Suche in Webarchiven
)
- ↑
Dietmar P. Berger, Rupert Engelhardt,
Roland Mertelsmann
:
Das Rote Buch: Hamatologie und Internistische Onkologie
. Huthig Jehle Rehm, 2013,
ISBN 978-3-609-51218-1
.
- ↑
Lukas Wagner:
Hamoglobin E ? Strukturvariante mit β-Thalassamiecharakter.
MVZ Labor 28,
abgerufen am 19. Oktober 2020
.
- ↑
Sichelzellanamie ? HBB ? Humangenetik ? Analysen-Spektrum ? Labor Lademannbogen.
Abgerufen am 10. April 2020
.
- ↑
2013 - 43 13.pdf.
In:
Epidemiologisches Bulletin
.
Abgerufen am 3. Februar 2014
.
- ↑
T. N. Williams, S. K. Obaro:
Sickle cell disease and malaria morbidity: a tale with two tails.
In:
Trends in Parasitology.
Band 27, Nummer 7, Juli 2011, S. 315?320.
doi:10.1016/j.pt.2011.02.004
- ↑
Joachim B. Kunz, Stephan Lobitz, Regine Grosse, Lena Oevermann, Dani Hakimeh:
Sickle cell disease in Germany: Results from a national registry
. In:
Pediatric Blood & Cancer
.
Band
67
,
Nr.
4
, 2020,
S.
e28130
,
doi
:
10.1002/pbc.28130
.
- ↑
J. I. Malowany, J. Butany:
Pathology of sickle cell disease.
In:
Seminars in Diagnostic Pathology.
Band 29, Nummer 1, Februar 2012, S. 49?55.
PMID 22372205
.
- ↑
A. U. Orjih, R. Chevli, C. D. Fitch:
Toxic heme in sickle cells: an explanation for death of malaria parasites
. In:
The American Journal of Tropical Medicine and Hygiene
.
Band
34
,
Nr.
2
, Marz 1985,
S.
223?227
,
PMID 3885769
.
- ↑
Skript der Harvard Medical School
(
Memento
vom 27. November 2011 im
Internet Archive
) (englisch). Abgerufen am 5. April 2024.
- ↑
I. Akinsheye, A. Alsultan, N. Solovieff, D. Ngo, C. T. Baldwin, P. Sebastiani, D. H. Chui, M. H. Steinberg:
Fetal hemoglobin in sickle cell anemia.
In:
Blood
.
Band 118, Nummer 1, Juli 2011, S. 19?27.
doi:10.1182/blood-2011-03-325258
.
PMID 21490337
.
PMC 3139383
(freier Volltext).
- ↑
O S Platt, S H Orkin, G Dover, G P Beardsley, B Miller:
Hydroxyurea enhances fetal hemoglobin production in sickle cell anemia
. In:
Journal of Clinical Investigation
.
Band
74
,
Nr.
2
, 1984,
S.
652?656
,
doi
:
10.1172/JCI111464
,
PMC 370519
(freier Volltext).
- ↑
Winfred C Wang, Russell E Ware, Scott T Miller, Rathi V Iyer, James F Casella:
Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG)
. In:
The Lancet
.
Band
377
,
Nr.
9778
, 14. Mai 2011,
S.
1663?1672
,
doi
:
10.1016/S0140-6736(11)60355-3
,
PMC 3133619
(freier Volltext).
- ↑
Ersi Voskaridou, Dimitrios Christoulas, Antonios Bilalis, Eleni Plata, Konstantinos Varvagiannis:
The effect of prolonged administration of hydroxyurea on morbidity and mortality in adult patients with sickle cell syndromes: results of a 17-year, single-center trial (LaSHS)
. In:
Blood
.
Band
115
,
Nr.
12
, 25. Marz 2010,
S.
2354?2363
,
doi
:
10.1182/blood-2009-05-221333
.
- ↑
Leon Tshilolo, George Tomlinson, Thomas N Williams, Brigida Santos, Peter Olupot-Olupot:
Hydroxyurea for Children with Sickle Cell Anemia in Sub-Saharan Africa
. In:
New England Journal of Medicine
.
Band
380
,
Nr.
2
, 10. Januar 2019,
S.
121?131
,
doi
:
10.1056/NEJMoa1813598
.
- ↑
C. Oringanje, E. Nemecek, O. Oniyangi:
Hematopoietic stem cell transplantation for people with sickle cell disease.
In:
The Cochrane database of systematic reviews.
Band 5, 2013, S. CD007001.
doi:10.1002/14651858.CD007001.pub3
- ↑
nature.com
- ↑
scinexx.de
- ↑
OXBRYTA- voxelotor tablet, film coated.
DailyMed
,
abgerufen am 7. November 2021
(englisch).
- ↑
FDA Approves First Gene Therapies to Treat Patients with Sickle Cell Disease
https://www.fda.gov/news-events/press-announcements/fda-approves-first-gene-therapies-treat-patients-sickle-cell-disease
Dieser Artikel behandelt ein Gesundheitsthema. Er dient
nicht
der Selbstdiagnose und ersetzt
nicht
eine Diagnose durch einen Arzt. Bitte hierzu den
Hinweis zu Gesundheitsthemen
beachten!